
Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Structure and Genomics of SARS-CoV-2
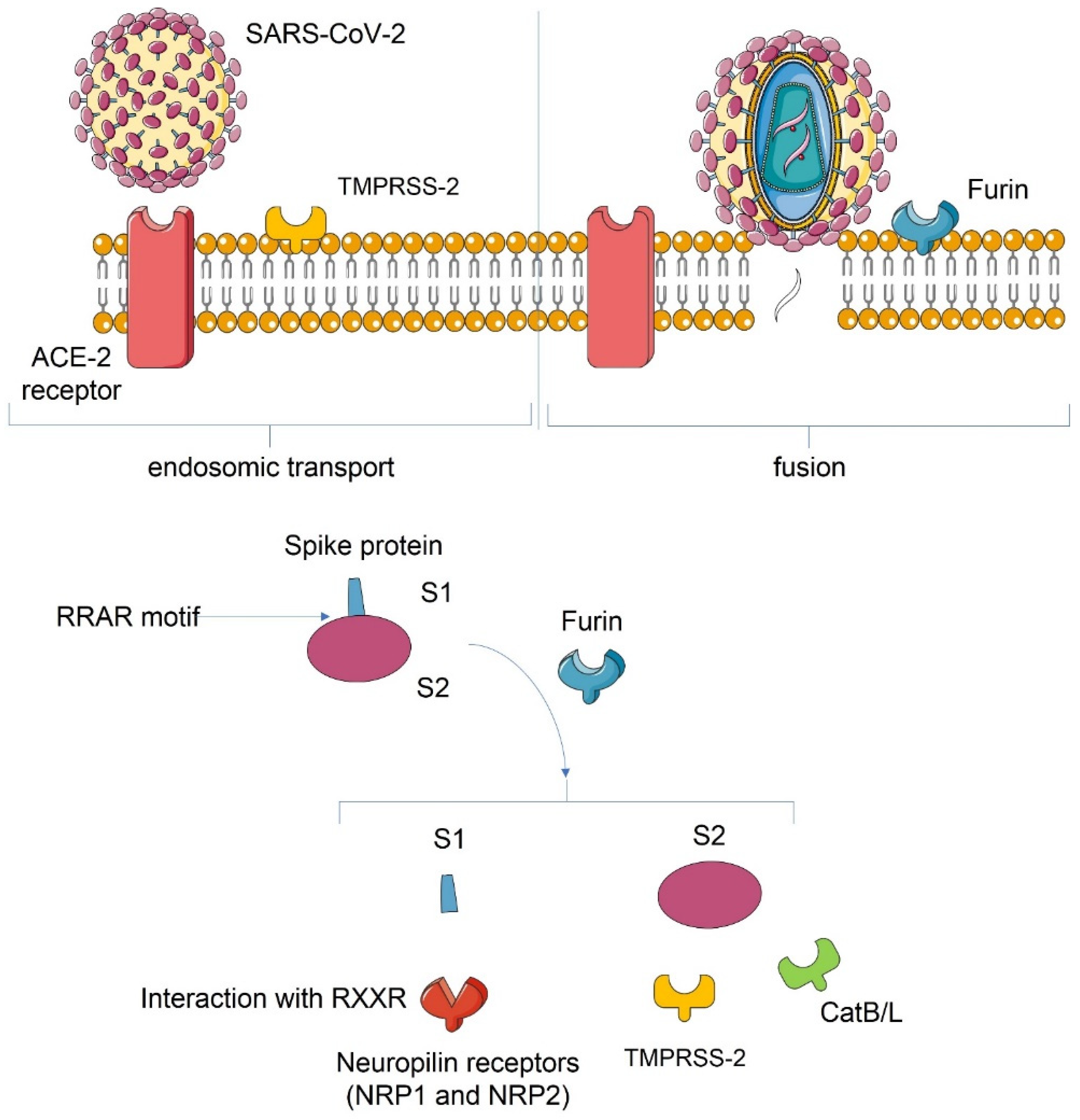
3. SARS-CoV-2 Host Interaction
3.1. Host Cell Interaction
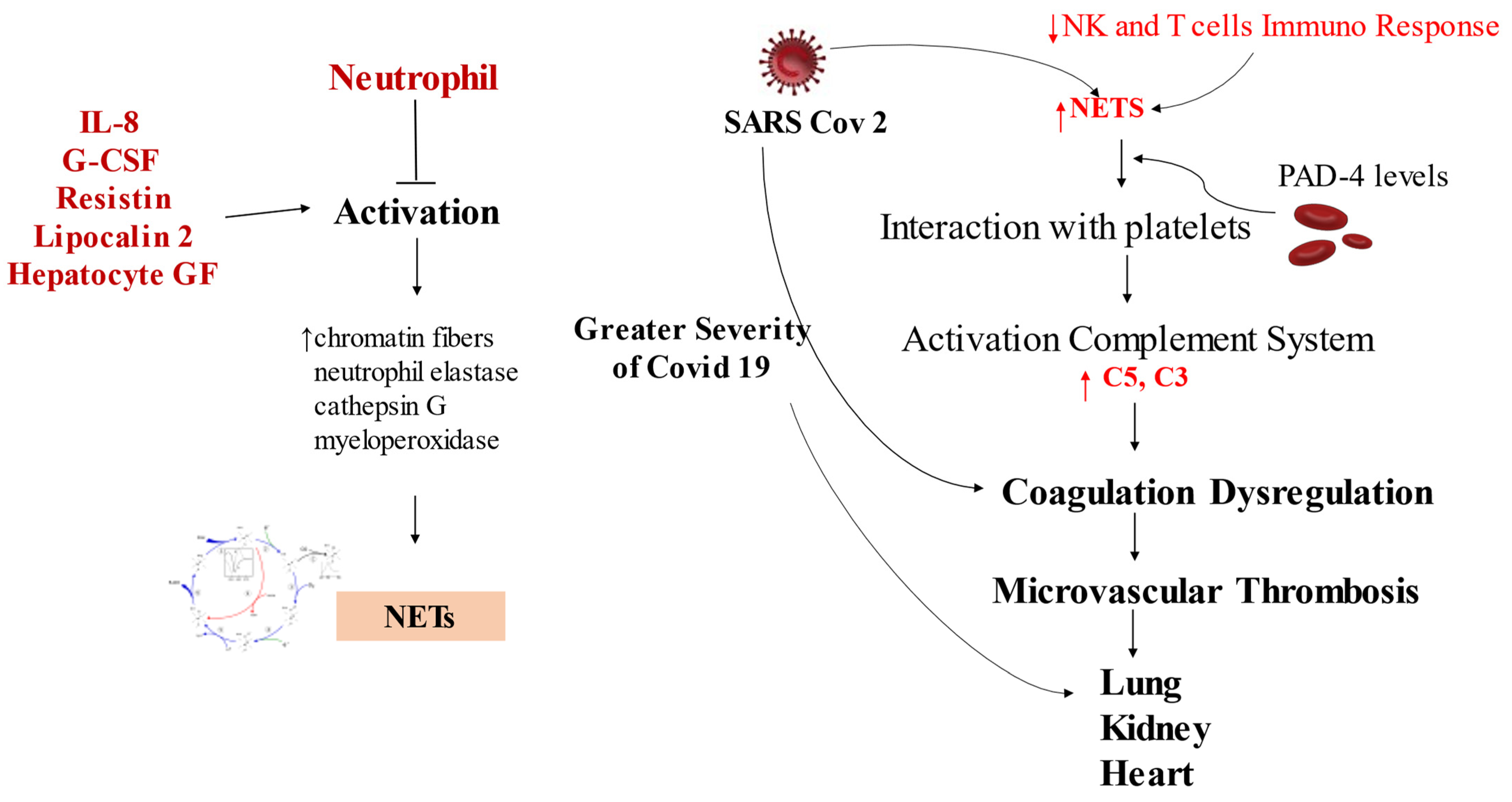
3.2. Host Response: How NETs Interfere
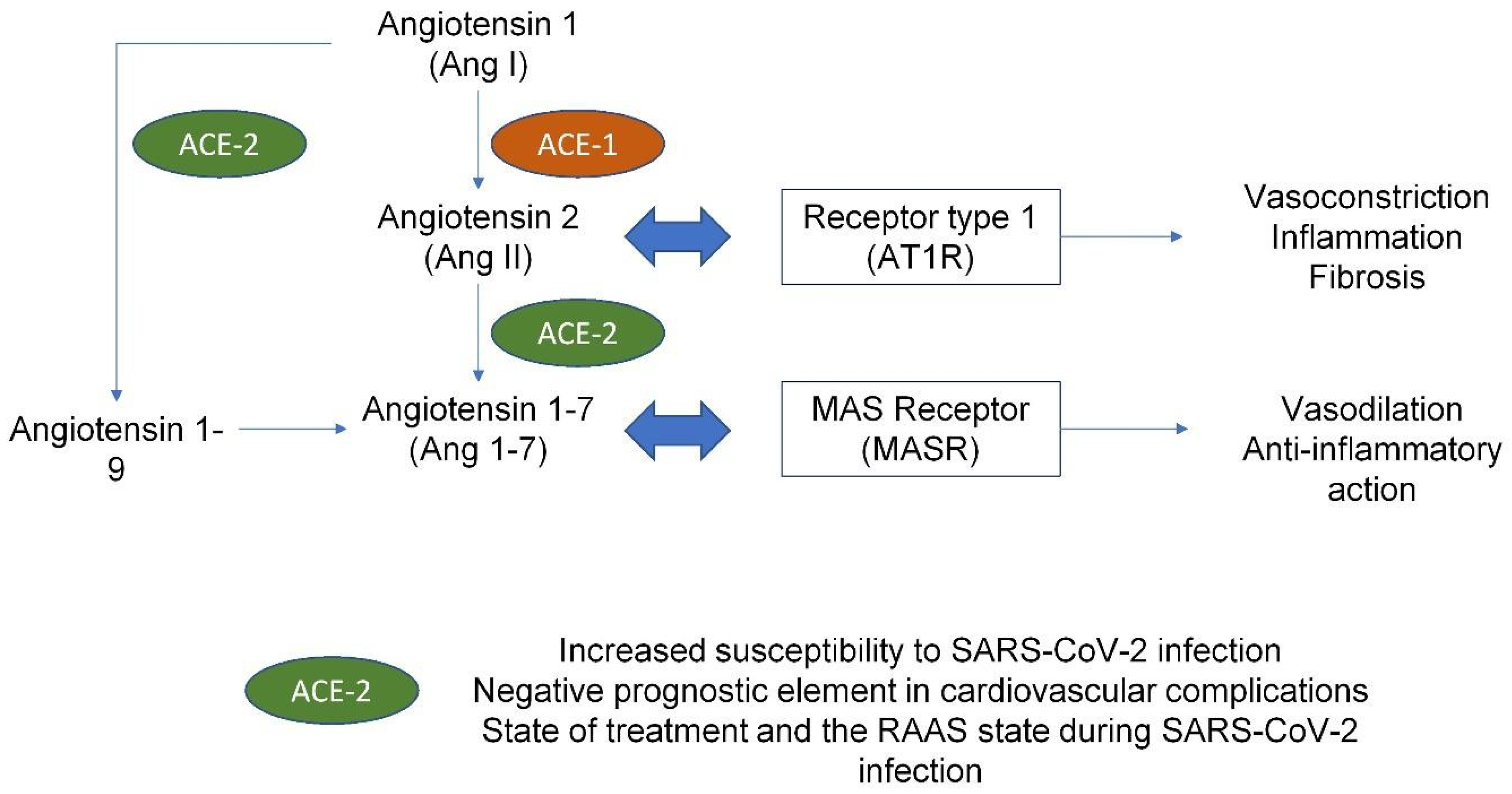
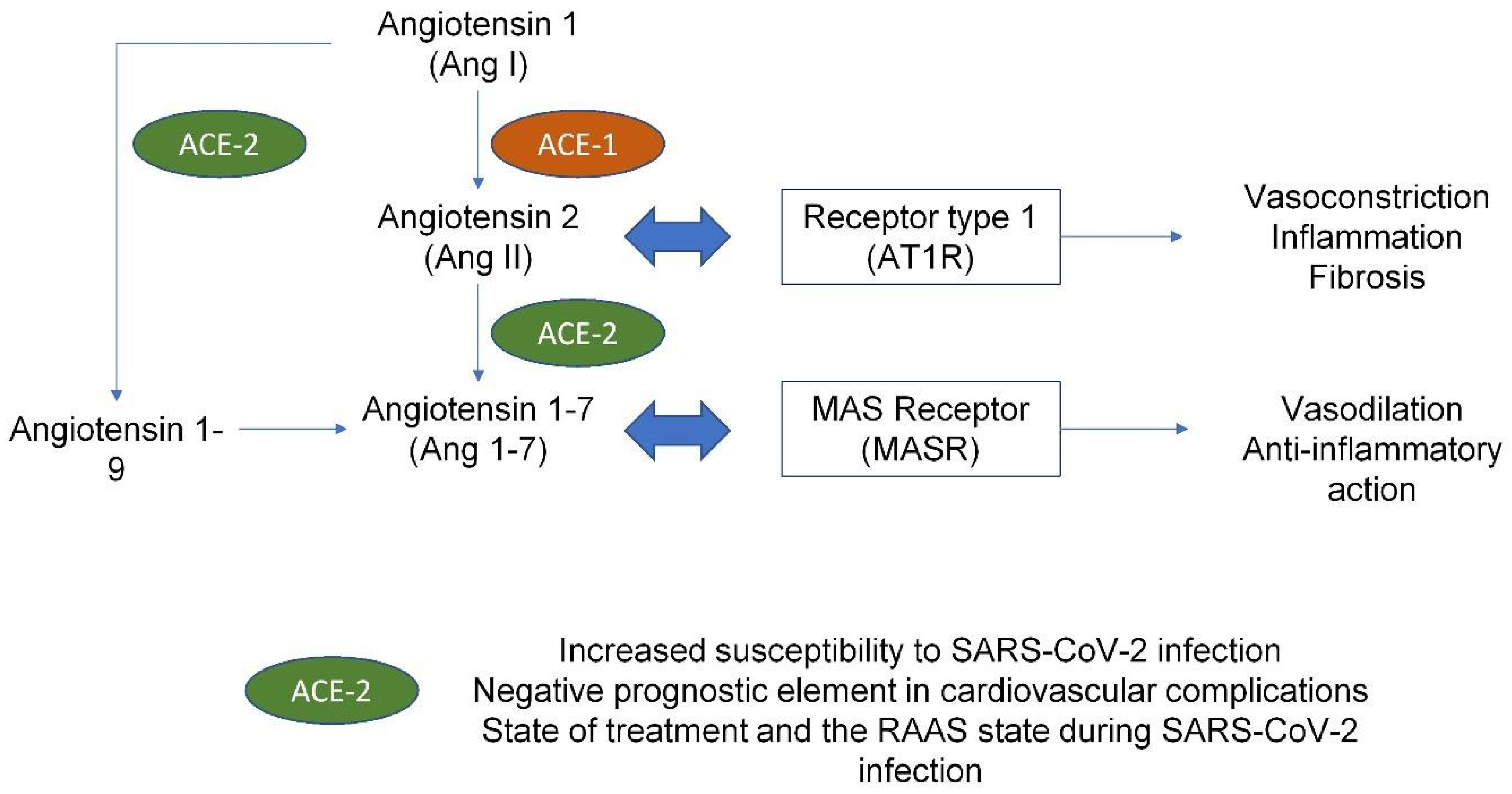
4. COVID Infection and Cardiovascular Implications
5. Dysregulation of Hemostasis Induced by SARS-CoV-2
5.1. Hypercoagulability in COVID Patients
5.2. The Role of NETs in Coronary Thrombus Formation
5.3. Antiphospholipid Syndrome and NETs: A Growing Concern for Microcirculation
5.4. The Role of Lipoprotein (a) and IL 6 in Arterial Thrombosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations/Acronyms
| Apo (a) | apolipoprotein (a) |
| ACE1 | angiotensin-converting enzyme 1 |
| ACE2 | angiotensin-converting enzyme 2 |
| ACEi ACE | inhibitors |
| aCL | anticardiolipin |
| ANA | antinuclear antibodies |
| ANCA | antineutrophil cytoplasmic antibodies |
| aPL | antiphospholipid |
| aPS/PT Ab | anti-phosphatidylserine/prothrombin autoantibodies |
| APS | antiphospholipid syndrome |
| ARDS | acute respiratory distress syndrome |
| AT1R | angiotensin type 1 receptor C1QBP |
| C1Q | binding protein |
| CatB/L | cysteine proteases cathepsin B/L |
| CendR | C-end rule |
| COVID-19 | Coronavirus disease 2019 |
| DIC | disseminated intravascular coagulation |
| DICER | Dipyridamole to Prevent Coronavirus Exacerbation of Respiratory Status |
| E | envelope |
| FDP | fibrinogen-derived peptide |
| G-CSF | granulocyte colony-stimulating factor |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| ICAM-1 | intercellular adhesion molecule 1 |
| ICU | intensive care unit |
| IL | interleukin |
| IP-10 | interferon-gamma-induced protein |
| ISG-15 | interferon-stimulated gene 15 |
| LDG | low-density granulocytes |
| LDL | low-density lipoprotein |
| Lp(a) | lipoprotein a |
| M | membrane protein |
| mAb | monoclonal antibody |
| MAM | domain in meprin, A5, receptor protein tyrosine phosphatase mu |
| MAPK | mitogen-associated protein kinase |
| MASP2 | mannose-binding protein-associated serine protease 2 |
| MAS-R | mitochondrial assembly receptor |
| MCP-1 | monocyte chemoattractant protein 1 |
| M-CSF | macrophage colony-stimulating factor |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MIP 1 | macrophage inflammatory protein 1 |
| N | nucleoprotein |
| NDG | normal-density granulocytes |
| NET | neutrophil extracellular trap |
| NRP1 | neuropilin receptor 1 |
| NRP2 | neuropilin receptor 2 |
| Nsps | nonstructural proteins |
| ORF | open reading frame |
| PAD | peptidyl arginine deaminase |
| PAI | platelet activator inhibitor |
| Pp | polyprotein |
| RAAS | renin–angiotensin–aldosterone system |
| RE | response element |
| rhACE2 | recombinant human |
| ACE2 S | spike |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| STEMI | ST-elevation myocardial infarction |
| TF | tissue factor |
| TFPI | tissue factor pathway inhibitor |
| TGF-β2 | transforming grow factor beta 2 |
| TMPRSS2 | transmembrane serine protease 2 |
| TNF | tumor necrosis factor |
| TRAP | thrombin receptor-activating peptide |
| TTSP | transmembrane serine protease |
| UTR | untranslated region |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VWF | von Willebrand factor |
| β2GPI | β2 I glycoprotein |
References
- Xiong, T.Y.; Redwood, S.; Prendergast, B.; Chen, M. Coronaviruses and the cardiovascular system: Acute and long-term im-plications. Eur. Heart J. 2020, 41, 1798–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kite, T.A.; Ludman, P.F.; Gale, C.P. International COVID-ACS Registry Investigators. International prospective registry of acute coronary syndromes in patients with COVID-19. J. Am. Coll. Cardiol. 2021, 77, 2466–2476. [Google Scholar] [CrossRef]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular considerations for patients, health care workers, and health systems during the coronavirus disease 2019 (COVID-19) pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.; Kennedy, K.F.; Imran, H.; Louis, D.W.; Shippey, E.; Poppas, A.; Wood, K.E.; Abbott, J.D.; Aronow, H.D. Association Between COVID-19 Diagnosis and In-Hospital Mortality in Patients Hospitalized with ST-Segment Elevation Myocardial Infarction. JAMA 2021, 326, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Madjid, M.; Safavi-Naeini, P.; Solomon, S.D.; Vardeny, O. Potential Effects of Coronaviruses on the Cardiovascular System: A Review. JAMA Cardiol. 2020, 5, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davogustto, G.E.; Clark, D.E.; Hardison, E.; Yanis, A.H.; Lowery, B.D.; Halasa, N.B.; Wells, Q.S. Characteristics Associated with Multisys- tem Inflammatory Syndrome Among Adults with SARS-CoV-2 Infection. JAMA Netw. Open 2021, 4, e2110323. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.Q.; Giustino, G.; Mahmood, K.; Oliveros, E.; Neibart, E.; Oloomi, M.; Moss, N.; Mitter, S.S.; Contreras, J.P.; Croft, L.; et al. Cardiogenic shock and hyperinflammatory syndrome in young males with COVID-19. Circ. Heart Fail. 2020, 13, e007485. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M. Overcoming COVID-19 Investigators; CDC COVID-19 Response Team.. Mul-tisystem inflammatory syndrome in U.S. children and adolescents. N. Engl. J. Med. 2020, 383, 334–346. [Google Scholar] [CrossRef]
- Dufort, E.M.; Koumans, E.H.; Chow, E.J.; Rosenthal, E.M.; Muse, A.; Rowlands, J.; Barranco, M.A.; Maxted, A.M.; Rosenberg, E.S.; Easton, D.; et al. New York State and Centers for Disease Control and Prevention Multisystem Inflam- matory Syndrome in Children Investigation Team. Multisystem inflammatory syndrome in children in New York State. N. Engl. J. Med. 2020, 383, 347–358. [Google Scholar] [CrossRef]
- Piazza, G.; Campia, U.; Hurwitz, S.; Snyder, J.E.; Rizzo, S.M.; Pfeferman, M.B.; Morrison, R.B.; Leiva, O.; Fanikos, J.; Nauffal, V.; et al. Registry of arterial and venous thromboembolic complications in patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in crit-ically ill COVID-19 patients. J. Thromb. Thrombolysis 2020, 50, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with res-piratory failure and coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection with SARS-CoV-2 in Confirmed COVID-19 Au- topsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Thierry, A.R.; Roch, B. Neutrophil extracellular traps and by-products play a key role in COVID-19: Pathogenesis, risk fac-tors, and therapy. J. Clin. Med. 2020, 9, 2942. [Google Scholar] [CrossRef]
- Mozzini, C.; Girelli, D. The role of neutrophil extracellular traps in COVID-19: Only an hypothesis or a potential new field of research? Thromb. Res. 2020, 191, 26–27. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Al-Hussaniy, H.A.; Al-Harcan, N.A.H.; Alexiou, A.; Batiha, G.E. Neutrophil Extracellu-lar Traps (NET) and COVID-19: A new frontiers for therapeutic modality. Int. Immunopharmacol. 2022, 104, 108516. [Google Scholar] [CrossRef]
- Singhal, A.; Yadav, S.; Chandra, T.; Mulay, S.R.; Gaikwad, A.N.; Kumar, S. An Imaging and Computational Algorithm for Efficient Identification and Quantification of Neutrophil Extracellular Traps. Cells 2022, 11, 191. [Google Scholar] [CrossRef]
- Blanch-Ruiz, M.A.; Ortega-Luna, R.; Gómez-García, G.; Martínez-Cuesta, M.Á.; Álvarez, Á. Role of Neutrophil Extracellular Traps in COVID-19 Progression: An Insight for Effective Treatment. Biomedicines 2021, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, A.; Coronado, M.J.; Hernández-Terciado, F.H.; Martín, P.; Royuela, A.; Ramil, E.; García, D.; Goicolea, J.; Del Trigo, M.; Ortega, J.; et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients with COVID-19 and Myocardial Infarction. JAMA Cardiol. 2020, 6, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Falasca, L.; Nardacci, R.; Colombo, D.; Lalle, E.; Di Caro, A.; Nicastri, E.; Antinori, A.; Petrosillo, N.; Marchioni, L.; Biava, G.; et al. Postmortem Findings in Italian Patients with COVID-19: A Descriptive Full Autopsy Study of Cases with and without Comorbidities. J. Infect. Dis. 2020, 222, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Xiang, X.; Zhang, W.; Yi, S.; Zhang, J.; Gu, X.; Xu, Y.; Huang, K.; Su, X.; Yu, B.; et al. Management and outcomes of patients with STEMI during the COVID-19 pandemic in China. J. Am. Coll. Cardiol. 2020, 76, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.S.; Gale, C.P.; Kinnaird, T.; Curzen, N.; Ludman, P.; Kontopantelis, E.; Wu, J.; Denwood, T.; Fazal, N.; Deanfield, J.; et al. Impact of COVID-19 on percutaneous coronary intervention for ST-elevationmyocardial infarction. Heart 2020, 106, 1805–1811. [Google Scholar] [CrossRef]
- Garcia, S.; Dehghani, P.; Grines, C. Society for Cardiac Angiography and Interventions, the Canadian Association of Interventional Cardiology, and the American College of Cardiology Interventional Council. Initial findings from the North American COVID-19 myocardial infarction registry. J. Am. Coll. Cardiol. 2021, 77, 1994–2003. [Google Scholar] [CrossRef]
- Choudry, F.A.; Hamshere, S.M.; Rathod, K.S.; Akhtar, M.M.; Archbold, R.A.; Guttmann, O.P.; Woldman, S.; Jain, A.K.; Knight, C.J.; Baumbach, A.; et al. High thrombus burden in patients with COVID-19 presenting with ST-segment elevationmyocardial infarction. J. Am. Coll. Cardiol. 2020, 76, 1168–1176. [Google Scholar] [CrossRef]
- Rodriguez-Leor, O.; Cid Alvarez, A.B.; Pérez de Prado, A.; Rossello, X.; Ojeda, S.; Serrador, A.; López-Palop, R.; Martin-Moreiras, J.; Rumoroso, J.R.; Cequier, A.; et al. In-hospital outcomes of COVID-19 ST-elevationmyocardial in-farction patients. EuroIntervention 2021, 16, 1426–1433. [Google Scholar] [CrossRef]
- Damluji, A.A.; Bandeen-Roche, K.; Berkower, C.; Boyd, C.M.; Al-Damluji, M.; Cohen, M.G.; Forman, D.E.; Chaudhary, R.; Gerstenblith, G.; Walston, J.D.; et al. Percutaneous Coronary Intervention in Older Patients with ST-Segment Elevation Myocardial Infarction and Cardiogenic Shock. J. Am. Coll. Cardiol. 2019, 73, 1890–1900. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with COVID-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2020, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients in-fected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Zandi, M.; Behboudi, E.; Soltani, S. Role of Glycoprotein Hemagglutinin-Esterase in COVID-19 Pathophysiology? Stem. Cell Rev. Rep. 2021, 17, 2359–2360. [Google Scholar] [CrossRef]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Thompson, R. Pandemic potential of 2019-nCoV. Lancet Infect. Dis. 2020, 20, 280. [Google Scholar] [CrossRef] [Green Version]
- Samidurai, A.; Das, A. Cardiovascular Complications Associated with COVID-19 and Potential Therapeutic—Strategies. Int. J. Mol. Sci. 2020, 21, 6790. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus fromWuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomencla-ture proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, K.; Liu, B.; Li, C.; Zhang, H.; Yu, T.; Qu, J.; Zhou, M.; Chen, L.; Meng, S.; Hu, Y.; et al. Eectiveness of convalescent plas-ma therapy in severe COVID-19 patients. Proc. Natl. Acad. Sci. USA 2020, 117, 9490–9496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin- 1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Carlos Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019- nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its me-tabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, M.; Uemura, K.; Sanaki, T.; Sato, A.; Hall, W.W.; Kariwa, H.; Orba, Y.; Sawa, H.; Sasaki, M. TMPRSS11D and TMPRSS13 activate the SARS-CoV-2 spike protein. Viruses 2021, 13, 384. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the effects of SARSCoV-2 spike muta-tion D614G on transmissibility and pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARSCoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Raeymaekers, V.; Van Berwaer, R.; Vandeput, J.; Marchand-Casas, I.; Thibaut, H.J.; Van Looveren, D.; Martens, K.; Hoffmann, M.; Maes, P.; et al. The SARS-CoV-2 and other human coronavirus spike proteins are fine-tuned towards temperature and proteases of the human airways. PLoS Pathog. 2021, 17, e1009500. [Google Scholar] [CrossRef]
- Meizlish, M.L.; Pine, A.B.; Bishai, J.D.; Goshua, G.; Nadelmann, E.R.; Simonov, M.; Chang, C.H.; Zhang, H.; Shallow, M.; Bahel, P.; et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021, 5, 1164–1177. [Google Scholar] [CrossRef]
- Schconrich, G.; Raftery, M.J. Neutrophil extracellular traps go viral. Front. Immunol. 2016, 7, 366. [Google Scholar]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martínez-Guzman, M.A.; Iniguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil extracellular traps and itsimplications in inflammation: An overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Knopf, J.; Maueroder, C.; Kienhofer, D.; Leppkes, M.; Herrmann, M. Neutrophils and neutrophil extracellular traps orchestrate initiation and resolution of inflammation. Clin. Exp. Rheumatol. 2016, 34, 6–8. [Google Scholar] [PubMed]
- Munoz, L.E.; Bilyy, R.; Biermann, M.H.C.; Kienh, D.; Maueroder, C.; Hahn, J.; Brauner, J.M.; Weidner, D.; Chen, J.; Scharin-Mehlmann, M.; et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, E5856–E5865. [Google Scholar] [CrossRef] [Green Version]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sin-clair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
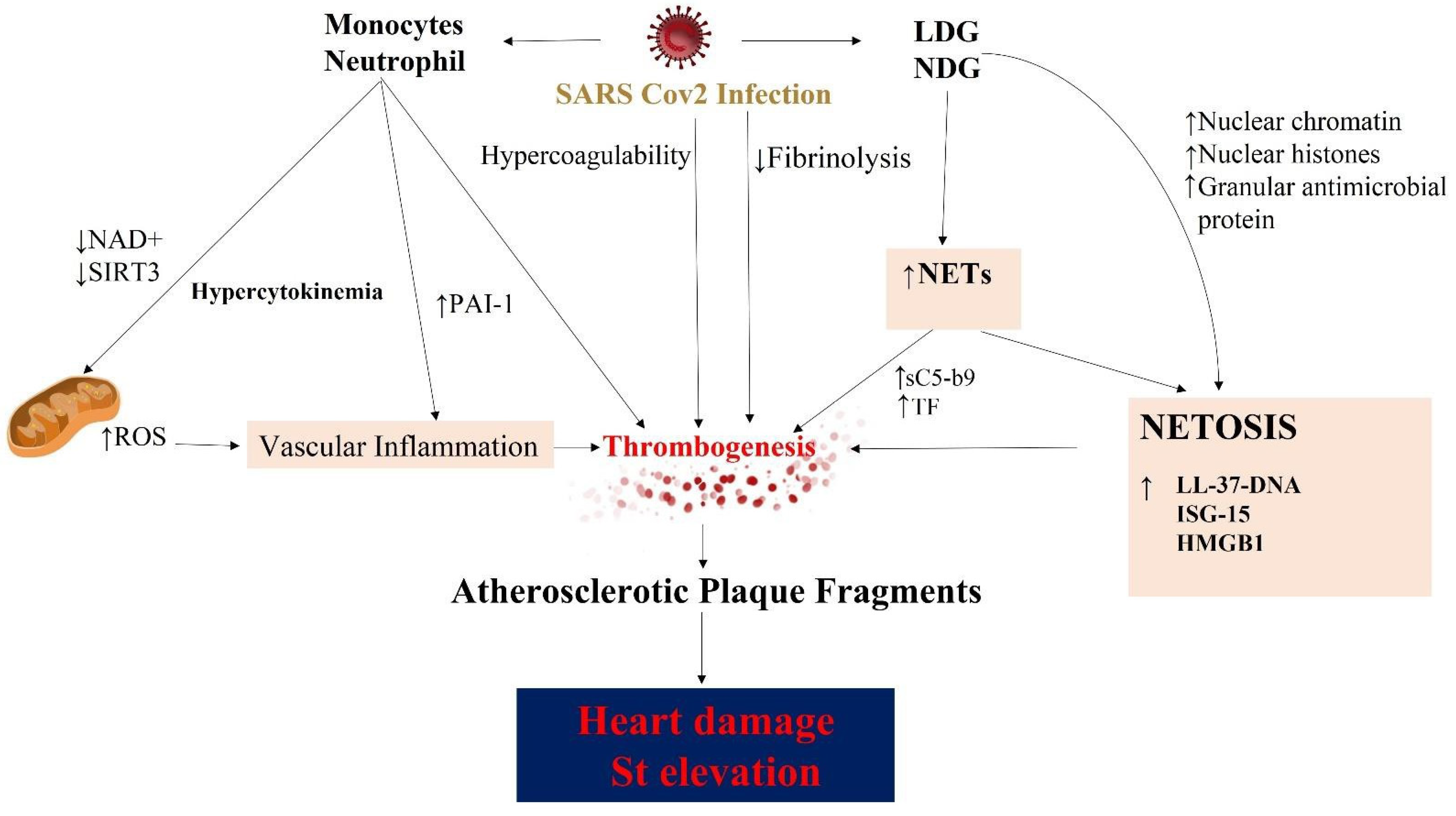
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps (NET) contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, M.; Knopf, J.; Naschberger, E.; Lindemann, A.; Singh, J.; Herrmann, I.; Stürzl, M.; Staats, L.; Mahajan, A.; Schauer, C.; et al. Vascular occlusion by neutrophil extracellular traps in COVID-19. EBioMedicine 2020, 58, 102925. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Pires da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Maccio, A.; Madeddu, C.; Caocci, G.; La Nasa, G. Multifactorial pathogenesis of COVID-19-related coagulopathy: Can de-fibrotide have a role in the early phases of coagulation disorders? J. Thromb. Haemost. 2020, 18, 3106–3108. [Google Scholar] [CrossRef] [PubMed]
- Price, L.C.; McCabe, C.; Garfield, B.; Wort, S.J. Thrombosis and COVID-19 pneumonia: The clot thickens! Eur. Respir. J. 2020, 56, 2001608. [Google Scholar] [CrossRef] [PubMed]
- Pujhari, S.; Paul, S.; Ahluwalia, J.; Rasgon, J.L. Clotting disorder in severe acute respiratory syndrome coronavirus 2. Rev. Med. Virol. 2020, 31, e2177. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive neutrophils and neutrophil extracellular traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and Individual Genetic Susceptibil-ity/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X- chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int. J. Mol. Sci. 2020, 21, 3474. [Google Scholar] [CrossRef] [PubMed]
- Zores, F.; Rebeaud, M.E. COVID and the Renin-Angiotensin System: Are Hypertension or Its Treatments Deleterious? Front. Cardiovasc. Med. 2020, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Olkowicz, M.; Chlopicki, S.; Smolenski, R.T. Perspectives for angiotensin profiling with liquid chromatography/mass spec-trometry to evaluate ACE/ACE2 balance in endothelial dysfunction and vascular pathologies. Pharmacol. Rep. 2015, 67, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raiza-da, M.K.; et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, D.; Italia, L.; Adamo, M.; Inciardi, R.M.; Lombardi, C.M.; Solomon, S.D.; Metra, M. COVID-19 and heart failure: From infection to inflammation and angiotensin II stimulation. Searching for evidence from a new disease. Eur. J. Heart Fail 2020, 22, 957–966. [Google Scholar] [CrossRef]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 recep-tor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.F.; Huang, F.Y.; Xiong, T.Y.; Liu, Q.; Chen, H.; Wang, H.; Huang, H.; Luo, Y.C.; Zhou, X.; Liu, Z.Y.; et al. Acute myocardial injury is common in patients with COVID-19 and impairs their prognosis. Heart 2020, 106, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Ng, C.Y.; Brook, R.H. Response to COVID-19 in Taiwan: Big Data Analytics, New Technology, and Proactive Test-ing. JAMA 2020, 323, 1341–1342. [Google Scholar] [CrossRef]
- Tersalvi, G.; Vicenzi, M.; Calabretta, D.; Biasco, L.; Pedrazzini, G.; Winterton, D. Elevated Troponin in Patients with Corona-virus Disease 2019: Possible Mechanisms. J. Card. Fail. 2020, 26, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
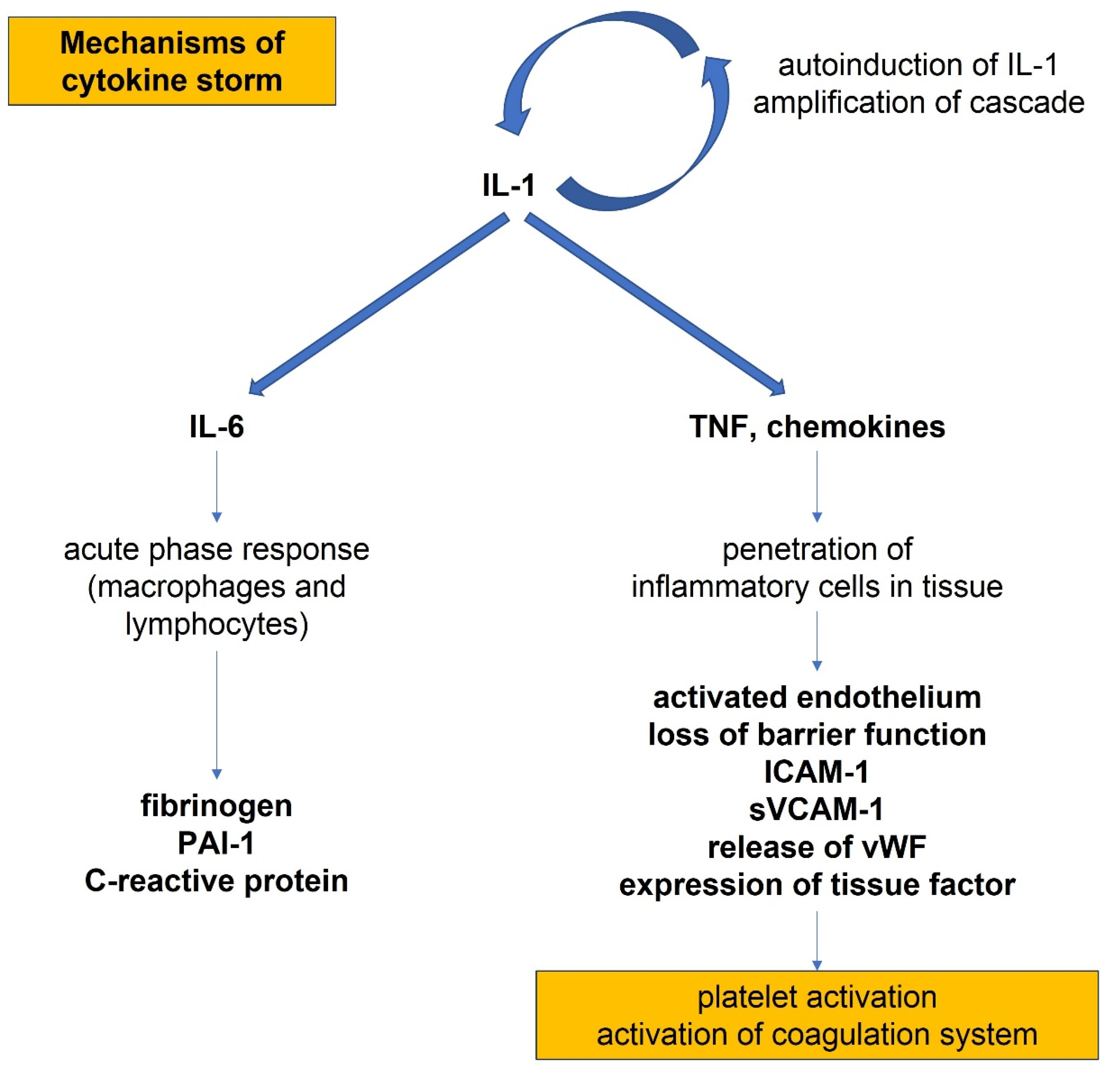
- Dinarello, C.A.; Ikejima, T.; Warner, S.J.C.; Orencole, S.F.; Lonnemann, G.; Cannon, J.G.; Libby, P. Interleukin-1 induces in-terleukin-1. I. Induction of circulating interleukin-1 in rabbits in vivo and in human mononuclear cells in vitro. J. Immunol. 1987, 139, 1902–1910. [Google Scholar] [PubMed]
- Warner, S.J.C.; Auger, K.R.; Libby, P. Interleukin-1 induces interleukin-1. II. Recombinant human interleukin-1 induces inter-leukin- 1 production by adult human vascular endothelial cells. J. Immunol. 1987, 139, 1911–1917. [Google Scholar]
- Warner, S.J.C.; Auger, K.R.; Libby, P. Human interleukin 1 induces interleukin 1 gene expression in human vascular smooth muscle cells. J. Exp. Med. 1987, 165, 1316–1331. [Google Scholar] [CrossRef]
- Wang, J.; Sica, A.; Peri, G.; Walter, S.; Martin-Padura, I.; Libby, P.; Ceska, M.; Lindley, I.; Colotta, F.; Mantovani, A. Expres-sion of monocyte chemotactic protein and interleukin-8 by cytokine-activated human vascular smooth muscle cells. Arterioscler. Thromb. 1991, 11, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; China Medical Treatment Expert Group for COVID-19; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Scientific and Standardization Committee on DIC, and the Scientific and Standardization Committee on Perioperative and Critical Care of the International Society on Thrombosis and Haemostasis, Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994. [Google Scholar] [PubMed] [Green Version]
- Iba, T.; Di Nisio, M.; Thachil, J.; Wada, H.; Asakura, H.; Sato, K.; Saitoh, D. A proposal of the modification of Japanese Soci-ety on Thrombosis and Hemostasis (JSTH) Disseminated Intravascular Coagulation (DIC) diagnostic criteria for sep-sis-associated DIC. Clin. Appl. Thromb. Hemost. 2017, 24, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paas-sen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.D.; Sacco, C.; Humanitas COVID-19 Task Force; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J. Thorac. Oncol. 2020, 15, 700–704. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
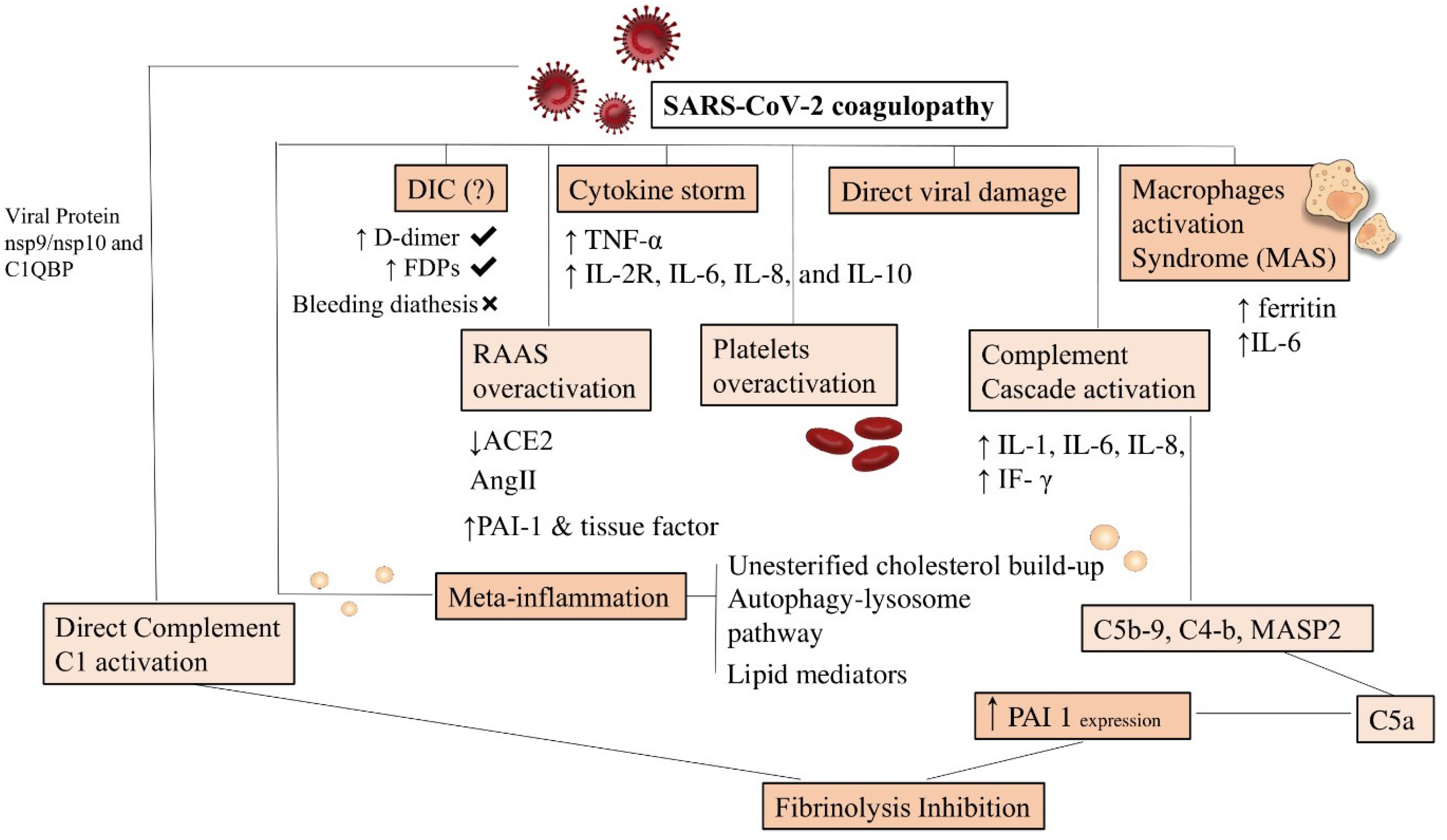
- Colling, M.E.; Kanthi, Y. COVID–19-associated coagulopathy: An exploration of mechanisms. Vasc. Med. 2020, 25, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zuo, Y.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Woodward, W.; Lezak, S.P.; Lugogo, N.L.; et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J. Leukoc. Biol. 2021, 109, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Argeting potential drivers of COVID 19: Neutrophil extracel-lular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C., Jr. Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. J. Am. Coll. Surg. 2020, 231, 193–203.e1. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lammle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Hanff, T.C.; Mohareb, A.M.; Giri, J.; Cohen, J.B.; Chirinos, J.A. Thrombosis in COVID-19. Am. J. Hematol. 2020, 95, 1578–1589. [Google Scholar] [CrossRef]
- Allnoch, L.; Beythien, G.; Leitzen, E.; Becker, K.; Kaup, F.; Stanelle-Bertram, S.; Schaumburg, B.; Kouassi, N.M.; Beck, S.; Zickler, M.; et al. Vascular Inflammation Is Associated with Loss of Aquaporin 1 Expres-sion on Endothelial Cells and Increased Fluid Leakage in SARS-CoV-2 Infected Golden Syrian Hamsters. Viruses 2021, 13, 639. [Google Scholar] [CrossRef]
- Kamel, M.H.; Yin, W.; Zavaro, C.; Francis, J.M.; Chitalia, V.C. Hyperthrombotic milieu in COVID-19 patients. Cells 2020, 9, 2392. [Google Scholar] [CrossRef]
- Endemann, D.H.; Schirin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef]
- Steel, J.; Luscher, T.F.; Tanner, F.C. Tissue factor in cardiovascular diseases: Molecular mechanisms and clinical implications. Circulation 2006, 113, 722–731. [Google Scholar]
- Butenas, S.; Orfeo, T.; Mann, K.G. Tissue factor activity and function in blood coagulation. Thromb. Res. 2008, 122, S42–S46. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopa-thy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wu, X.; Zheng, X.; Luo, S.; Xu, S.; Weng, J. Targeting inflammation and cytokine storm in COVID-19. Pharm. Res. 2020, 159, 105051. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Wright, D.J.M. Prevention of the cytokine storm in COVID-19. Lancet Infect. Dis. 2020, 21, 25–26. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Szotowski, B.; Antoniak, S.; Poller, W.; Schultheiss, H.P.; Rauch, U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ. Res. 2005, 96, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Levi, M.; Scully, M. How I treat disseminated intravascular coagulation. Blood 2018, 131, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Wilk, C.M. Coronaviruses hijack the complement system. Nat. Rev. Immunol. 2020, 20, 350. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associat-ed microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-host interactome and prote-omic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med. N. Y. 2021, 2, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Handtke, S.; Thiele, T. Large and small platelets-(When) do they dier? J. Thromb. Haemost. 2020, 18, 1256–1267. [Google Scholar] [CrossRef]
- Handtke, S.; Steil, L.; Palankar, R.; Conrad, J.; Cauhan, S.; Kraus, L.; Ferrara, M.; Dhople, V.; Wesche, J.; Volker, U.; et al. Role of Platelet Size Revisited-Function and Protein Composition of Large and Small Platelets. Thromb. Haemost. 2019, 119, 407–420. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in pa-tients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Laube, B.; Abu Abed, U.; Goosmann, C.; Zychlinsky, A. Neutrophil extracellular traps: How to generate and visualize them. J. Vis. Exp. 2010, 24, 1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Ruiz, J.; Absalón-Aguilar, A.; Nuñez-Aguirre, M.; Pérez-Fragoso, A.; Carrillo-Vázquez, D.A.; Maravillas-Montero, J.L.; Mejía-Domínguez, N.R.; Llorente, L.; Alcalá-Carmona, B.; Lira-Luna, J.; et al. Neutrophil Extracellular Traps Contribute to COVID-19 Hyperinflammation and Humoral Autoimmunity. Cells 2021, 10, 2545. [Google Scholar] [CrossRef]
- Langseth, M.S.; Helseth, R.; Ritschel, V.; Hansen, C.H.; Andersen, G.Ø.; Eritsland, J.; Halvorsen, S.; Fagerland, M.W.; Solheim, S.; Arnesen, H.; et al. Double-stranded DNA and NET components in relation to clinical outcome after ST-elevation myocardial infarction. Sci. Rep. 2020, 10, 5007. [Google Scholar] [CrossRef]
- Madison, J.A.; Duarte-García, A.; Zuo, Y.; Knight, J.S. Treatment of thrombotic antiphospholipid syndrome in adults and children. Curr. Opin. Rheumatol. 2020, 32, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Shi, H.; Li, C.; Knight, J.S. Antiphospholipid syndrome: A clinical perspective. Chin. Med. J. 2020, 133, 929–940. [Google Scholar] [CrossRef]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphos-pholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef]
- Kazzaz, N.M.; McCune, W.J.; Knight, J.S. Treatment of catastrophic antiphospholipid syndrome. Curr. Opin. Rheumatol. 2016, 28, 218–227. [Google Scholar] [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Shi, H.; Zheng, H.; Yin, Y.F.; Hu, Q.Y.; Teng, J.L.; Sun, Y.; Liu, H.L.; Cheng, X.B.; Ye, J.N.; Su, Y.T.; et al. Antiphosphatidylserine/prothrombin antibodies (aPS/PT) as potential diagnostic markers and risk predictors of venous thrombosis and obstetric complications in antiphospho-lipid syndrome. Clin. Chem. Lab. Med. 2018, 56, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Devreese, K.M.J.; Linskens, E.A.; Benoit, D.; Peperstraete, H. Antiphospholipid antibodies in patients with COVID-19: A rel-evant observation? J. Thromb. Haemost. 2020, 18, 2191–2201. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef] [PubMed]
- Gatto, M.; Perricone, C.; Tonello, M.; Bistoni, O.; Cattelan, A.M.; Bursi, R.; Cafaro, G.; De Robertis, E.; Mencacci, A.; Bozza, S.; et al. Frequency and clinical correlates of an-tiphospholipid antibodies arising in patients with SARS-CoV-2 infection: Findings from a multicentre study on 122 cases. Clin. Exp. Rheumatol. 2020, 38, 754–759. [Google Scholar] [PubMed]
- Siguret, V.; Voicu, S.; Neuwirth, M.; Delrue, M.; Gayat, E.; Stépanian, A.; Mégarbane, B. Are antiphospholipid antibodies associated with thrombotic complications in critically ill COVID-19 patients? Thromb. Res. 2020, 195, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Zhang, Y.; Zhang, S.; Qin, X.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; et al. Brief report: Anti-phospholipid antibodies in critically ill pa-tients with Coronavirus Disease 2019 (COVID-19). Arthritis Rheumatol. 2020, 72, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Borghi, M.O.; Beltagy, A.; Garrafa, E.; Curreli, D.; Cecchini, G.; Bodio, C.; Grossi, C.; Blengino, S.; Tincani, A.; Franceschini, F.; et al. Prevalence, specificity, and clinical association of anti-phospholipid antibodies in COVID-19 patients: Are the antibodies really guilty? medRxiv 2020, 16. [Google Scholar] [CrossRef]
- Abdel-Wahab, N.; Talathi, S.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E. Risk of developing antiphospholipid antibodies fol-lowing viral infection: A systematic review and metaanalysis. Lupus 2018, 27, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Asherson, R.A.; Cervera, R. ‘Primary’, ‘secondary’ and other variants of the antiphospholipid syndrome. Lupus 1994, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, N.; Lopez-Olivo, M.A.; Pinto-Patarroyo, G.P.; Suarez-Almazor, M.E. Systematic review of case reports of an-tiphospholipid syndrome following infection. Lupus 2016, 25, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.; Anjum, S. Coronavirus Disease 2019 (COVID-19) infection associated with antiphospholipid antibodies and four- extremity deep vein thrombosis in a previously healthy female. Cureus 2020, 12, e8408. [Google Scholar] [CrossRef]
- Zuo, Y.; Zuo, M.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Shi, H.; Woodard, W.; Lezak, S.P.; Lugogo, N.L.; Knight, J.S.; et al. Neutrophil extracellular traps and thrombosis in COVID-19. J. Thromb. Thrombolysis 2021, 51, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; De Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the dark-NET of neu-trophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Navaz, S.A.; Hoy, C.K.; Harbaugh, A.; Gockman, K.; Zuo, M.; Madison, J.A.; Shi, H.; Kanthi, Y.; et al. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. JCI Insight 2021, 6, e150111. [Google Scholar] [CrossRef] [PubMed]
- Szturmowicz, M.; Demkow, U. Neutrophil Extracellular Traps (NET) in Severe SARS-CoV-2 Lung Disease. Int. J. Mol. Sci. 2021, 22, 8854. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic auto-antibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Shi, H.; Zuo, Y.; Navaz, S.; Harbaugh, A.; Hoy, C.K.; Gandhi, A.A.; Sule, G.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; et al. Endothelial cell-activating antibodies in COVID-19. medRxiv 2021, 2021.01.18.21250041. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F. Incertitude Pathophysiology and Management During the First Phase of the COVID-19 Pandemic. Ann. Thorac. Surg. 2022, 113, 693. [Google Scholar] [CrossRef] [PubMed]
- COVID Surg Collaborative; GlobalSurg Collaborative. Timing of surgery following SARS-CoV-2 infection: An international prospective cohort study. Anaesthesia 2021, 76, 748–758. [Google Scholar] [CrossRef] [PubMed]
- COVID Surg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 infection and venous thromboembolism after surgery: An international prospective cohort study. Anaesthesia 2022, 77, 28–39. [Google Scholar] [CrossRef]
- Nappi, F.; Iervolino, A.; Avtaar Singh, S.S. Thromboembolic Complications of SARS-CoV-2 and Metabolic Derangements: Suggestions from Clinical Practice Evidence to Causative Agents. Metabolites 2021, 11, 341. [Google Scholar] [CrossRef]
- COVID Surg Collaborative; GlobalSurg Collaborative. SARS-CoV-2 vaccination modelling for safe surgery to save lives: Data from an international prospective cohort study. Br. J. Surg. 2021, 108, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- COVID Surg Collaborative; GlobalSurg Collaborative. Effects of pre-operative isolation on postoperative pulmonary compli-cations after elective surgery: An international prospective cohort study. Anaesthesia 2021, 76, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.T.; Goepp, M.; Rossi, A.G.; Yao, C. Non-steroidal anti-inflammatory drugs, prostaglandins, and COVID-19. Br. J. Pharmacol. 2020, 177, 4899–4920. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.Y.; et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 2020, 10, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S. Dipyridamole to Prevent Coronavirus Exacerbation of Respiratory Status (DICER) in COVID-19 (DICER). Available online: https://clinicaltrials.gov/ct2/show/NCT04391179 (accessed on 25 January 2022).
- Buechler, C.; Ullrich, H.; Ritter, M.; Porsch-Oezcueruemez, M.; Lackner, K.J.; Barlage, S.; Friedrich, S.O.; Kostner, G.M.; Schmitz, G. Lipoprotein (a) up-regulates the expression of the plasminogen activator inhibitor 2 in human blood monocytes. Blood 2001, 97, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Caplice, N.M.; Panetta, C.; Peterson, T.E.; Kleppe, L.S.; Mueske, C.S.; Kostner, G.M.; Broze, G.J., Jr.; Simari, R.D. Lipoprotein (a) binds and inacti-vates tissue factor pathway inhibitor: A novel link between lipoproteins and thrombosis. Blood 2001, 98, 2980–2987. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Isshiki, H.; Sugita, T.; Tanabe, O.; Kinoshita, S.; Nishio, Y.; Nakajima, T.; Hirano, T.; Kishimoto, T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990, 9, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.P.; Clarke, J.G.; Lindahl, G.E.; Liu, A.C.; Zysow, B.R.; Meer, K.; Schwartz, K.; Lawn, R.M. 5′ control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proc. Natl. Acad. Sci. USA 1993, 90, 1369–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, N.; Schulte, D.M.; Türk, K.; Freitag-Wolf, S.; Hampe, J.; Zeuner, R.; Schröder, J.O.; Gouni-Berthold, I.; Berthold, H.K.; Krone, W. IL-6 blockade by monoclonal antibodies in-hibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J. Lipid. Res. 2015, 56, 1034–1042. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, P.M.; Gorby, L.K.; Stroes, E.S.; Kastelein, J.P.; Davidson, M.; Tsimikas, S. Lipoprotein(a) and Its Potential Associa-tion with Thrombosis and Inflammation in COVID-19: A Testable Hypothesis. Curr. Atheroscler. Rep. 2020, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Horváth, L.; Császár, A.; Falus, A.; Dieplinger, H.; Horváth, A.; Puskás, E.; Halm, G.; Bányai, A.; Pálóczi, K.; László, E.; et al. IL-6 and lipoprotein(a) [LP(a)] concentra-tions are related only in patients with high APO(a) isoforms in monoclonal gammopathy. Cytokine 2002, 18, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Schultz, O.; Oberhauser, F.; Saech, J.; Rubbert-Roth, A.; Hahn, M.; Krone, W.; Laudes, M. Effects of inhibition of interleukin-6 sig-nalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS ONE 2010, 5, e14328. [Google Scholar] [CrossRef] [PubMed]
- Berthold, H.K.; Laudes, M.; Krone, W.; Gouni-Berthold, I. Association between the interleukin-6 promoter polymorphism -174G/C and serum lipoprotein(a) concentrations in humans. PLoS ONE 2011, 6, e24719. [Google Scholar] [CrossRef] [Green Version]
- Giacinto, O.; Satriano, U.; Nenna, A.; Spadaccio, C.; Lusini, M.; Mastroianni, C.; Nappi, F.; Chello, M. Inflammatory Re-sponse and Endothelial Dysfunction Following Cardiopulmonary Bypass: Pathophysiology and Pharmacological Targets. Recent. Pat. Inflamm. Allergy Drug. Discov. 2019, 13, 158–173. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Strains | Description |
|---|---|
| L strain and the similar ORF8-L84S Strain | originated in Wuhan, China |
| S strain | mutation of ORF8, L84S |
| V strain | variant of ORF3a-coding protein NS3, G251V |
| G strain | mutation in spike protein, D614G |
| GH strain | mutations in spike protein, D614G and ORF3a, Q57H |
| GR strain | mutation in nucleocapsid gene, RG203KR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Giacinto, O.; Ellouze, O.; Nenna, A.; Avtaar Singh, S.S.; Chello, M.; Bouzguenda, A.; Copie, X. Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review. Biomedicines 2022, 10, 702. https://doi.org/10.3390/biomedicines10030702
Nappi F, Giacinto O, Ellouze O, Nenna A, Avtaar Singh SS, Chello M, Bouzguenda A, Copie X. Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review. Biomedicines. 2022; 10(3):702. https://doi.org/10.3390/biomedicines10030702
Chicago/Turabian StyleNappi, Francesco, Omar Giacinto, Omar Ellouze, Antonio Nenna, Sanjeet Singh Avtaar Singh, Massimo Chello, Assine Bouzguenda, and Xavier Copie. 2022. "Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review" Biomedicines 10, no. 3: 702. https://doi.org/10.3390/biomedicines10030702
APA StyleNappi, F., Giacinto, O., Ellouze, O., Nenna, A., Avtaar Singh, S. S., Chello, M., Bouzguenda, A., & Copie, X. (2022). Association between COVID-19 Diagnosis and Coronary Artery Thrombosis: A Narrative Review. Biomedicines, 10(3), 702. https://doi.org/10.3390/biomedicines10030702