
Combined HP 13C Pyruvate and 13C-Glucose Fluxomic as a Potential Marker of Response to Targeted Therapies in YUMM1.7 Melanoma Xenografts

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tumor Models
2.2. Animal Treatment
2.3. Tissue Fixation and Freezing
2.4. Homogenisation of Tumors
2.5. Western Blot
2.6. Hyperpolarized 13C-MRS
2.7. 13C-MRS
2.8. Statistical Analysis
3. Results
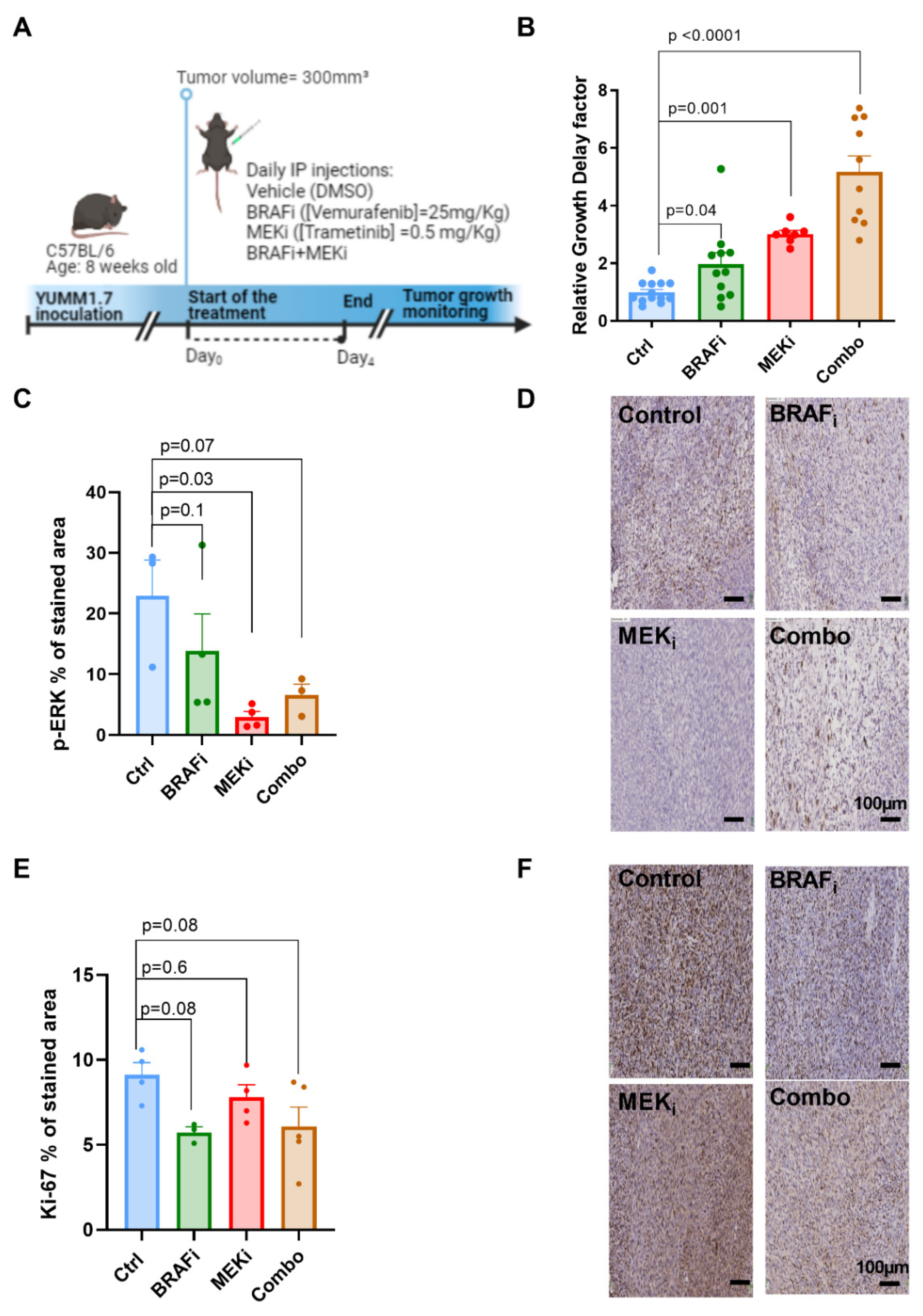
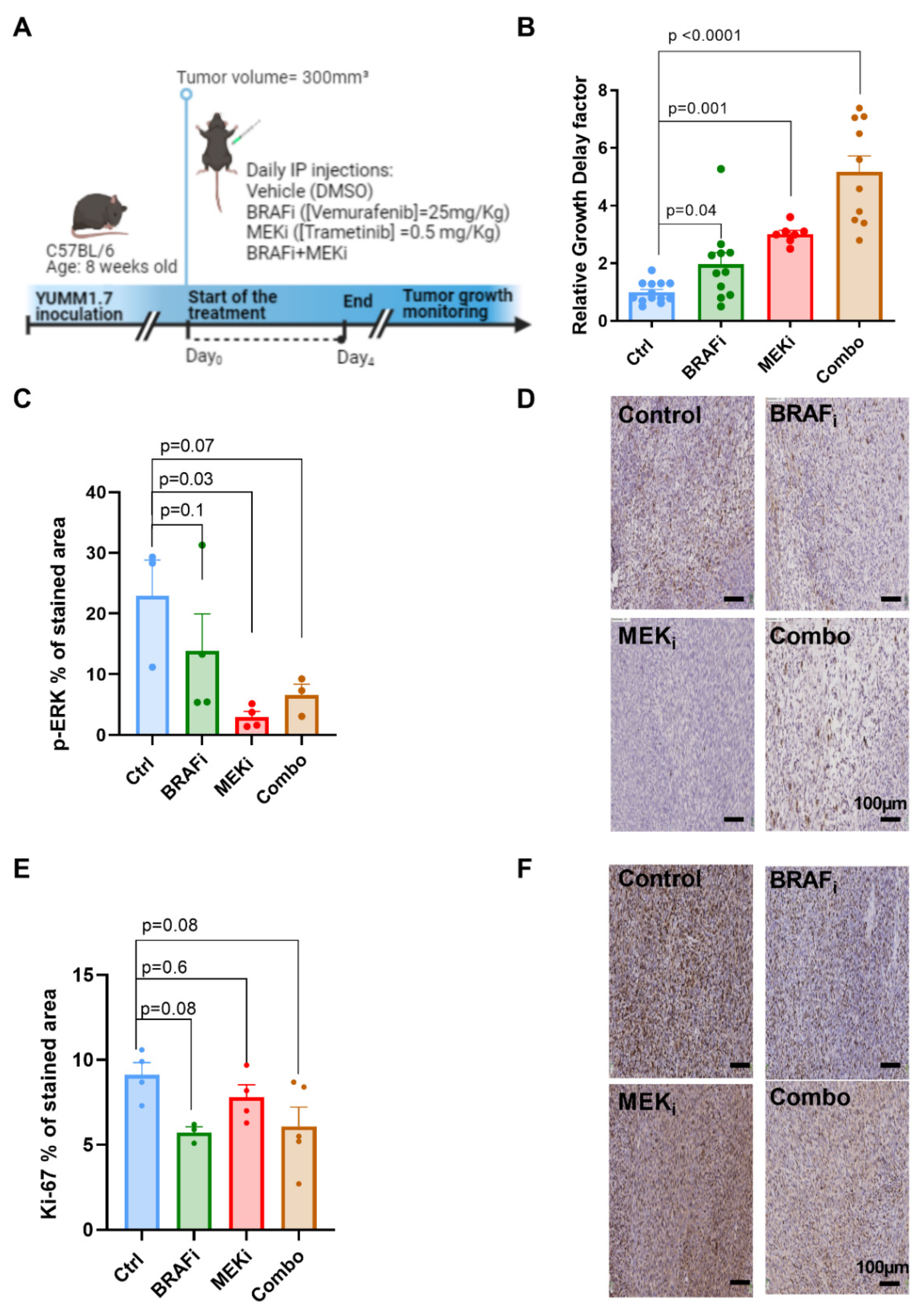
3.1. Combined BRAF and MEK Inhibition Delays Tumor Growth to a Larger Extent Than BRAF or MEK Inhibition Alone in Syngeneic YUMM1.7 Melanoma Xenografts
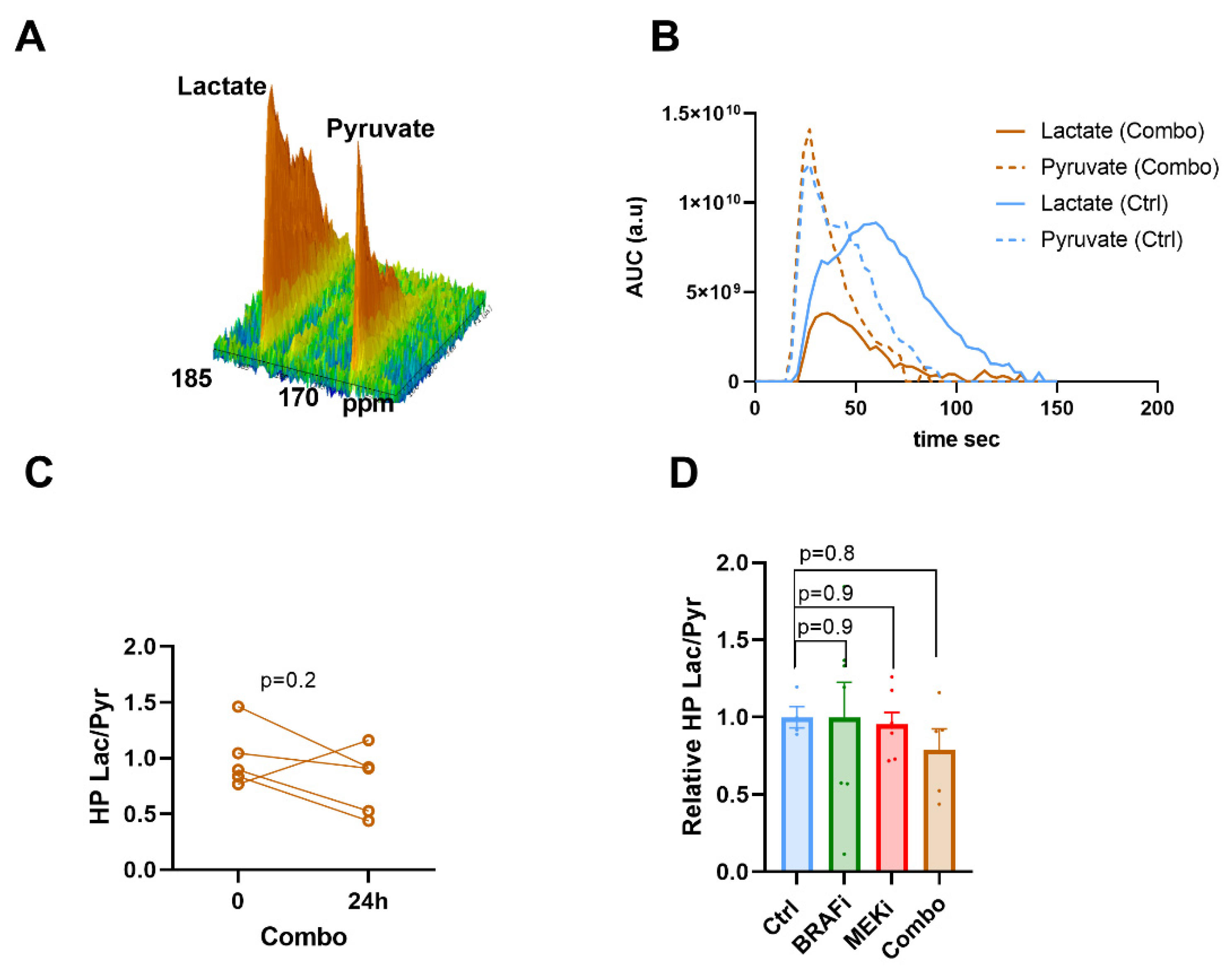
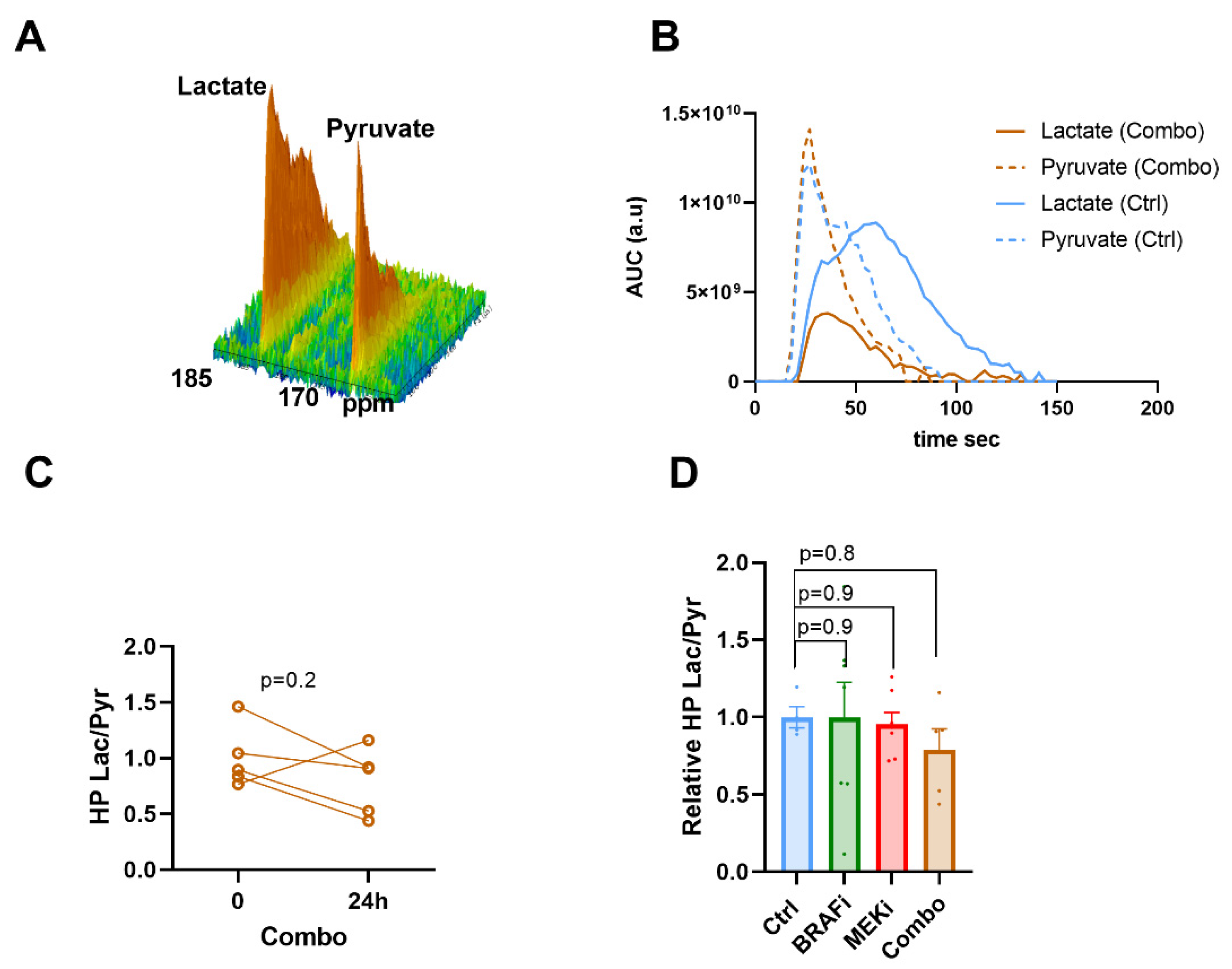
3.2. Hyperpolarized 13C Pyruvate -Lactate Exchange Is not Modified in Response to BRAF and/or MEK Inhibition
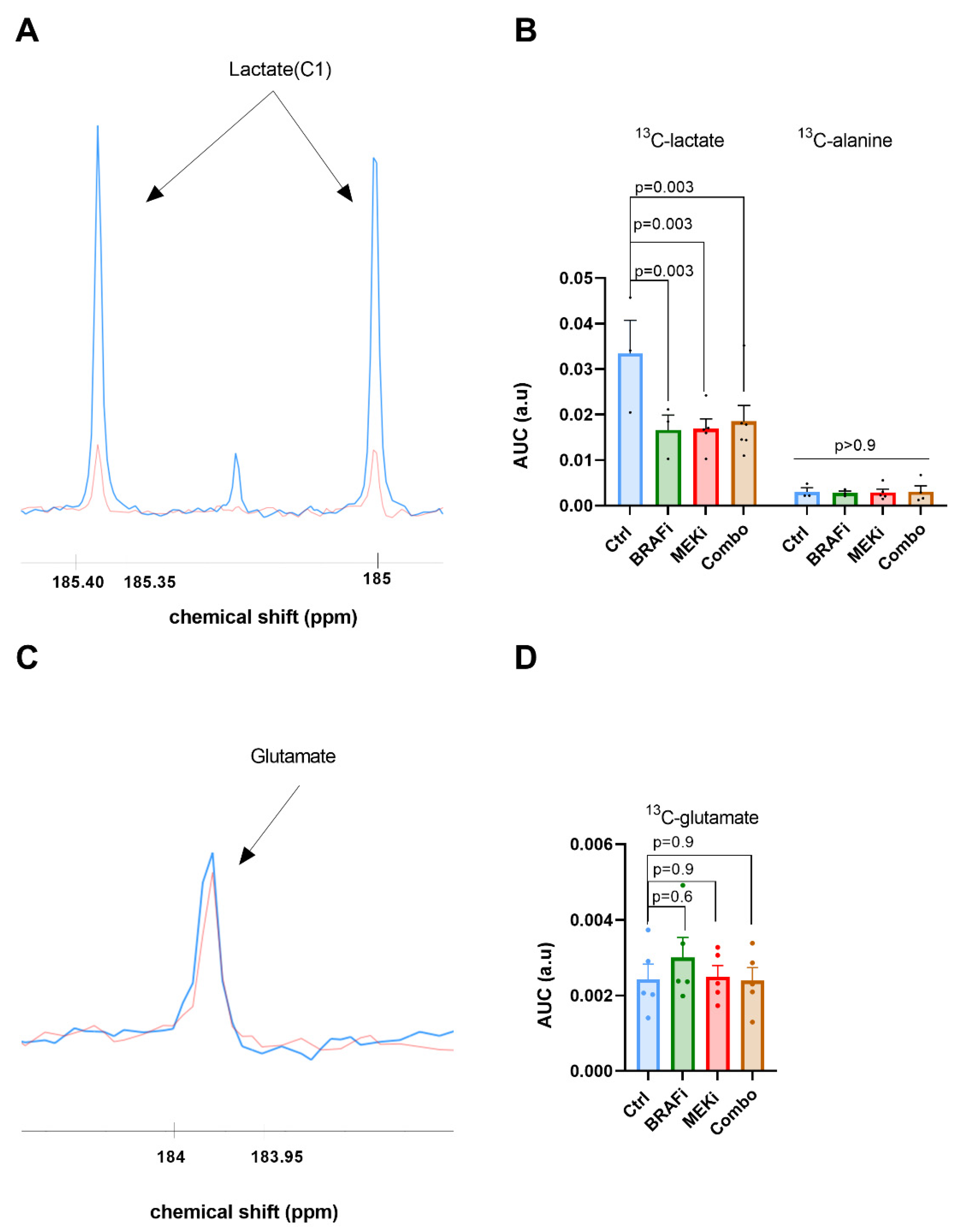
3.3. In Vivo 13C Glucose Fluxomic, and Not 13C Glutamine, Detects Metabolic Changes in Response to BRAF and/or MEK Inhibition
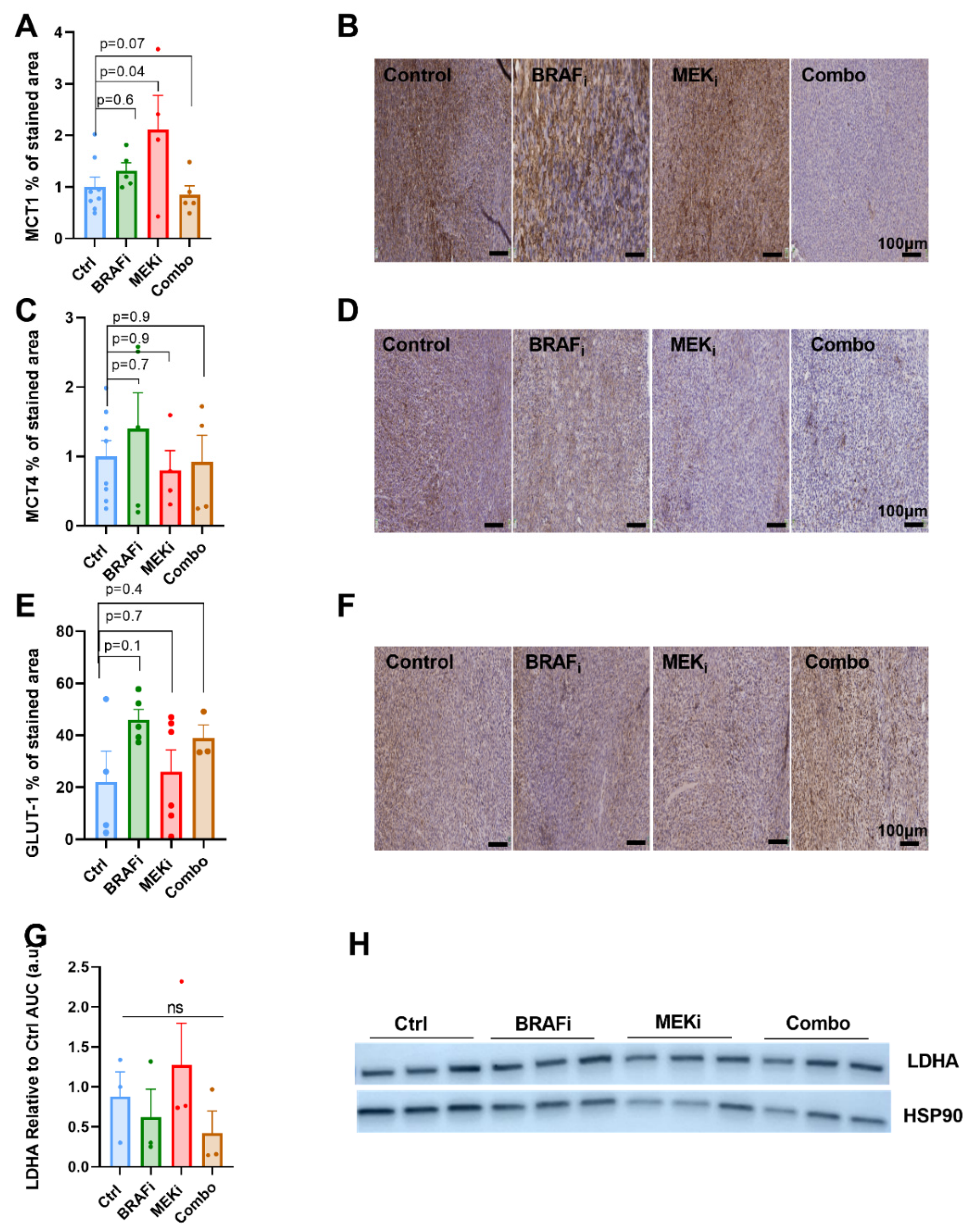
3.4. The Expression of Metabolic Transporters or LDH-A Are Not Modified in Response to BRAF or MEK Inhibition in YUMM1.7 Xenografts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma Treatment in Review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic Effects and Mechanisms of Resistance to Vemurafenib in Patients with Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas Acquire Resistance to B-RAF(V600E) Inhibition by RTK or N-RAS Upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/MTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Prieto, P.A.; Reuben, A.; Cooper, Z.A.; Wargo, J.A. Targeted Therapies Combined With Immune Checkpoint Therapy. Cancer J. Sudbury Mass 2016, 22, 138–146. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted Agents and Immunotherapies: Optimizing Outcomes in Melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, Y.N.V.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of MTORC1/2 Overcomes Resistance to MAPK Pathway Inhibitors Mediated by PGC1α and Oxidative Phosphorylation in Melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus Ipilimumab or Nivolumab Alone versus Ipilimumab Alone in Advanced Melanoma (CheckMate 067): 4-Year Outcomes of a Multicentre, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Brummer, C.; Faerber, S.; Bruss, C.; Blank, C.; Lacroix, R.; Haferkamp, S.; Herr, W.; Kreutz, M.; Renner, K. Metabolic Targeting Synergizes with MAPK Inhibition and Delays Drug Resistance in Melanoma. Cancer Lett. 2019, 442, 453–463. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: Beyond the Warburg Effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Goni, T.; Miniotis, M.F.; Wantuch, S.; Parkes, H.G.; Marais, R.; Workman, P.; Leach, M.O.; Beloueche-Babari, M. The BRAF Inhibitor Vemurafenib Activates Mitochondrial Metabolism and Inhibits Hyperpolarized Pyruvate-Lactate Exchange in BRAF-Mutant Human Melanoma Cells. Mol. Cancer Ther. 2016, 15, 2987–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-Mutant Melanoma to BRAF Inhibition Is Mediated by a Network of Transcriptional Regulators of Glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Chaneton, B.; Smith, M.; Van Den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF Inhibitors Induces Glutamine Dependency in Melanoma Cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef]
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S.; et al. Vemurafenib Resistance Reprograms Melanoma Cells towards Glutamine Dependence. J. Transl. Med. 2015, 13, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose Metabolism and NRF2 Coordinate the Antioxidant Response in Melanoma Resistant to MAPK Inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudchadkar, R.; Paraiso, K.H.T.; Smalley, K.S.M. Targeting Mutant BRAF in Melanoma: Current Status and Future Development of Combination Therapy Strategies. Cancer J. Sudbury Mass 2012, 18, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buder-Bakhaya, K.; Hassel, J.C. Biomarkers for Clinical Benefit of Immune Checkpoint Inhibitor Treatment-A Review From the Melanoma Perspective and Beyond. Front. Immunol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Almuhaideb, A.; Papathanasiou, N.; Bomanji, J. 18F-FDG PET/CT Imaging in Oncology. Ann. Saudi Med. 2011, 31, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Witney, T.H.; Kettunen, M.I.; Day, S.E.; Hu, D.; Neves, A.A.; Gallagher, F.A.; Fulton, S.M.; Brindle, K.M. A Comparison between Radiolabeled Fluorodeoxyglucose Uptake and Hyperpolarized (13)C-Labeled Pyruvate Utilization as Methods for Detecting Tumor Response to Treatment. Neopl. N. Y. N 2009, 11, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Baudy, A.R.; Dogan, T.; Flores-Mercado, J.E.; Hoeflich, K.P.; Su, F.; van Bruggen, N.; Williams, S.-P. FDG-PET Is a Good Biomarker of Both Early Response and Acquired Resistance in BRAFV600 Mutant Melanomas Treated with Vemurafenib and the MEK Inhibitor GDC-0973. EJNMMI Res. 2012, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- McArthur, G.A.; Puzanov, I.; Amaravadi, R.; Ribas, A.; Chapman, P.; Kim, K.B.; Sosman, J.A.; Lee, R.J.; Nolop, K.; Flaherty, K.T.; et al. Marked, Homogeneous, and Early [18F]Fluorodeoxyglucose-Positron Emission Tomography Responses to Vemurafenib in BRAF-Mutant Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.J.; Vigneron, D.; Kurhanewicz, J.; Chen, A.; Bok, R.; Hurd, R. DNP-Hyperpolarized 13C Magnetic Resonance Metabolic Imaging for Cancer Applications. Appl. Magn. Reson. 2008, 34, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Mäurer, J.; Tosi, C.; Hierholzer, J.; Bier, J.; Felix, J.; Lagalla, R.; Midiri, M.; Finazzo, M. The assessment of skin tumors with magnetic resonance with high resolution and a paramagnetic contrast medium. Radiol. Med. 1998, 95, 563–566. [Google Scholar]
- Shulman, R.G.; Brown, T.R.; Ugurbil, K.; Ogawa, S.; Cohen, S.M.; den Hollander, J.A. Cellular Applications of 31P and 13C Nuclear Magnetic Resonance. Science 1979, 205, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Golman, K.; Zandt, R.I.; Lerche, M.; Pehrson, R.; Ardenkjaer-Larsen, J.H. Metabolic Imaging by Hyperpolarized 13C Magnetic Resonance Imaging for in Vivo Tumor Diagnosis. Cancer Res. 2006, 66, 10855–10860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurhanewicz, J.; Vigneron, D.B.; Brindle, K.; Chekmenev, E.Y.; Comment, A.; Cunningham, C.H.; Deberardinis, R.J.; Green, G.G.; Leach, M.O.; Rajan, S.S.; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neopl. N. Y. N 2011, 13, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Falck Miniotis, M.; Arunan, V.; Eykyn, T.R.; Marais, R.; Workman, P.; Leach, M.O.; Beloueche-Babari, M. MEK1/2 Inhibition Decreases Lactate in BRAF-Driven Human Cancer Cells. Cancer Res. 2013, 73, 4039–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acciardo, S.; Mignion, L.; Lacomblez, E.; Schoonjans, C.; Joudiou, N.; Gourgue, F.; Bouzin, C.; Baurain, J.-F.; Gallez, B.; Jordan, B.F. Metabolic Imaging Using Hyperpolarized 13 C-Pyruvate to Assess Sensitivity to the B-Raf Inhibitor Vemurafenib in Melanoma Cells and Xenografts. J. Cell. Mol. Med. 2020, 24, 1934–1944. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Kremer, D.M.; Huang, H.; Breitkopf, S.B.; Ben-Sahra, I.; Manning, B.D.; Lyssiotis, C.A.; Asara, J.M. Ex Vivo and in Vivo Stable Isotope Labelling of Central Carbon Metabolism and Related Pathways with Analysis by LC-MS/MS. Nat. Protoc. 2019, 14, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Yan, J.; Fan, T.W.-M. 13C Tracer Studies of Metabolism in Mouse Tumor Xenografts. Bio-Protoc. 2015, 5, 4–5. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK Inhibition with PD-1 Blockade Immunotherapy in BRAF-Mutant Melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and Trametinib versus Dabrafenib and Placebo for Val600 BRAF-Mutant Melanoma: A Multicentre, Double-Blind, Phase 3 Randomised Controlled Trial. Lancet Lond. Engl. 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Meeth, K.; Wang, J.; Micevic, G.; Damsky, W.; Bosenberg, M.W. The YUMM Lines: A Series of Congenic Mouse Melanoma Cell Lines with Defined Genetic Alterations. Pigm. Cell Melanoma Res. 2016, 29, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Borst, A.; Adam, C.; Becker, T.M.; Motschenbacher, S.; Windhövel, S.; Hufnagel, A.L.; Houben, R.; Meierjohann, S. Vemurafenib Induces Senescence Features in Melanoma Cells. J. Invest. Dermatol. 2013, 133, 1601–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.F.; Jiang, C.C.; Kiejda, K.A.; Gillespie, S.; Zhang, X.D.; Hersey, P. Apoptosis Induction in Human Melanoma Cells by Inhibition of MEK Is Caspase-Independent and Mediated by the Bcl-2 Family Members PUMA, Bim, and Mcl-1. Clin. Cancer Res. 2007, 13, 4934–4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurhanewicz, J.; Vigneron, D.B.; Ardenkjaer-Larsen, J.H.; Bankson, J.A.; Brindle, K.; Cunningham, C.H.; Gallagher, F.A.; Keshari, K.R.; Kjaer, A.; Laustsen, C.; et al. Hyperpolarized 13C MRI: Path to Clinical Translation in Oncology. Neopl. N. Y. N 2019, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]Glucose in Human Brain Tumors in Vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Gammon, S.; Zacharias, N.M.; Liu, T.; Salzillo, T.; Xi, Y.; Wang, J.; Bhattacharya, P.; Piwnica-Worms, D. Hyperpolarized [1- 13 C]Pyruvate-to-[1- 13 C]Lactate Conversion Is Rate-Limited by Monocarboxylate Transporter-1 in the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 22378–22389. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farah, C.; Neveu, M.-A.; Yelek, C.; Bouzin, C.; Gallez, B.; Baurain, J.-F.; Mignion, L.; Jordan, B.F. Combined HP 13C Pyruvate and 13C-Glucose Fluxomic as a Potential Marker of Response to Targeted Therapies in YUMM1.7 Melanoma Xenografts. Biomedicines 2022, 10, 717. https://doi.org/10.3390/biomedicines10030717
Farah C, Neveu M-A, Yelek C, Bouzin C, Gallez B, Baurain J-F, Mignion L, Jordan BF. Combined HP 13C Pyruvate and 13C-Glucose Fluxomic as a Potential Marker of Response to Targeted Therapies in YUMM1.7 Melanoma Xenografts. Biomedicines. 2022; 10(3):717. https://doi.org/10.3390/biomedicines10030717
Chicago/Turabian StyleFarah, Chantale, Marie-Aline Neveu, Caner Yelek, Caroline Bouzin, Bernard Gallez, Jean-François Baurain, Lionel Mignion, and Bénédicte F. Jordan. 2022. "Combined HP 13C Pyruvate and 13C-Glucose Fluxomic as a Potential Marker of Response to Targeted Therapies in YUMM1.7 Melanoma Xenografts" Biomedicines 10, no. 3: 717. https://doi.org/10.3390/biomedicines10030717
APA StyleFarah, C., Neveu, M.-A., Yelek, C., Bouzin, C., Gallez, B., Baurain, J.-F., Mignion, L., & Jordan, B. F. (2022). Combined HP 13C Pyruvate and 13C-Glucose Fluxomic as a Potential Marker of Response to Targeted Therapies in YUMM1.7 Melanoma Xenografts. Biomedicines, 10(3), 717. https://doi.org/10.3390/biomedicines10030717