
Anti-Inflammatory CDGSH Iron-Sulfur Domain 2: A Biomarker of Central Nervous System Insult in Cellular, Animal Models and Patients

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Patients
2.3. Animals
2.4. Spinal Cord Hemisection
2.5. Collection of CSF from Rats with SCI
2.6. Cytokine Antibody Array Assay
2.7. Experimental Groups of the Mouse SCI Model
2.8. Administration of Anti-CISD2 Neutralizing Antibody in Mice
2.9. Histology, Immunohistochemistry, and Cell Count
2.10. Cell Lines
2.11. Experimental Grouping of In Vitro Microglial Cells
2.12. RNA Interference
2.13. RT-qPCR Analysis
2.14. Immunoblotting
2.15. Detection of NF-κB p65 Activity
2.16. ELISA
2.17. Statistical Analyses
3. Results
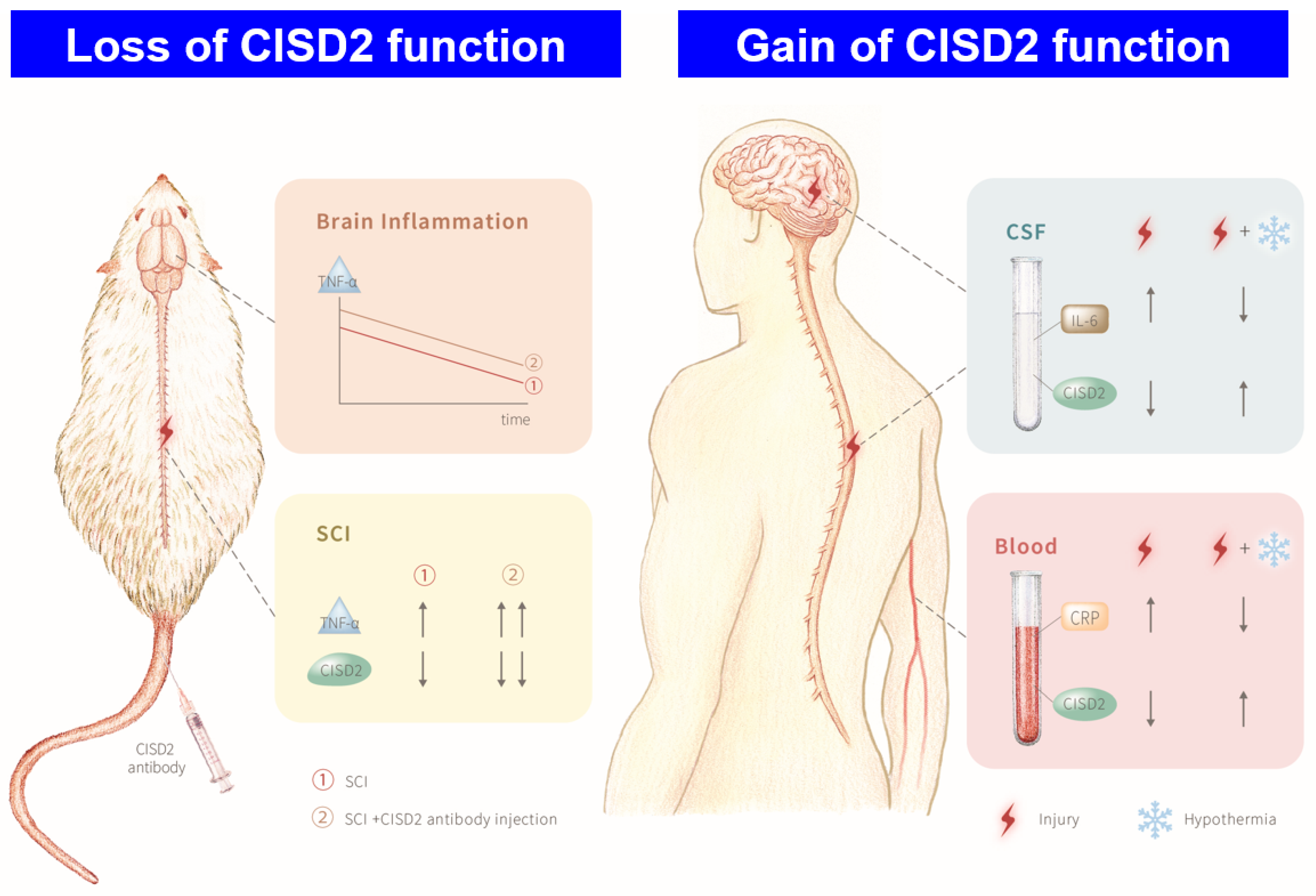
3.1. Plasma and CSF Levels of CISD2 Were Decreased in Patients with CNS Insult and Negatively Correlated with the Severity of the Injury
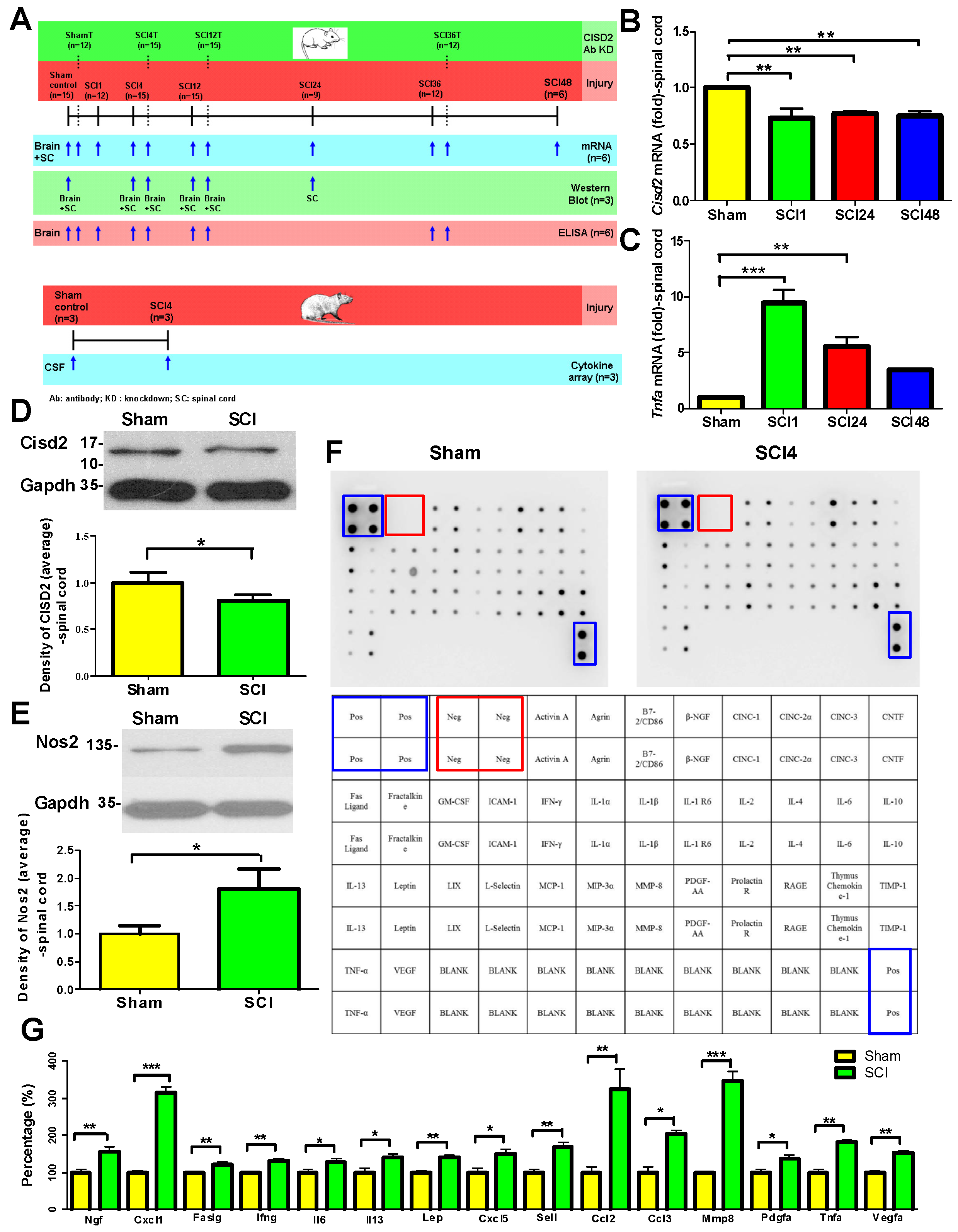
3.2. Mouse Model of SCI Exhibited Inflammation in the Injured Spine and the Brain
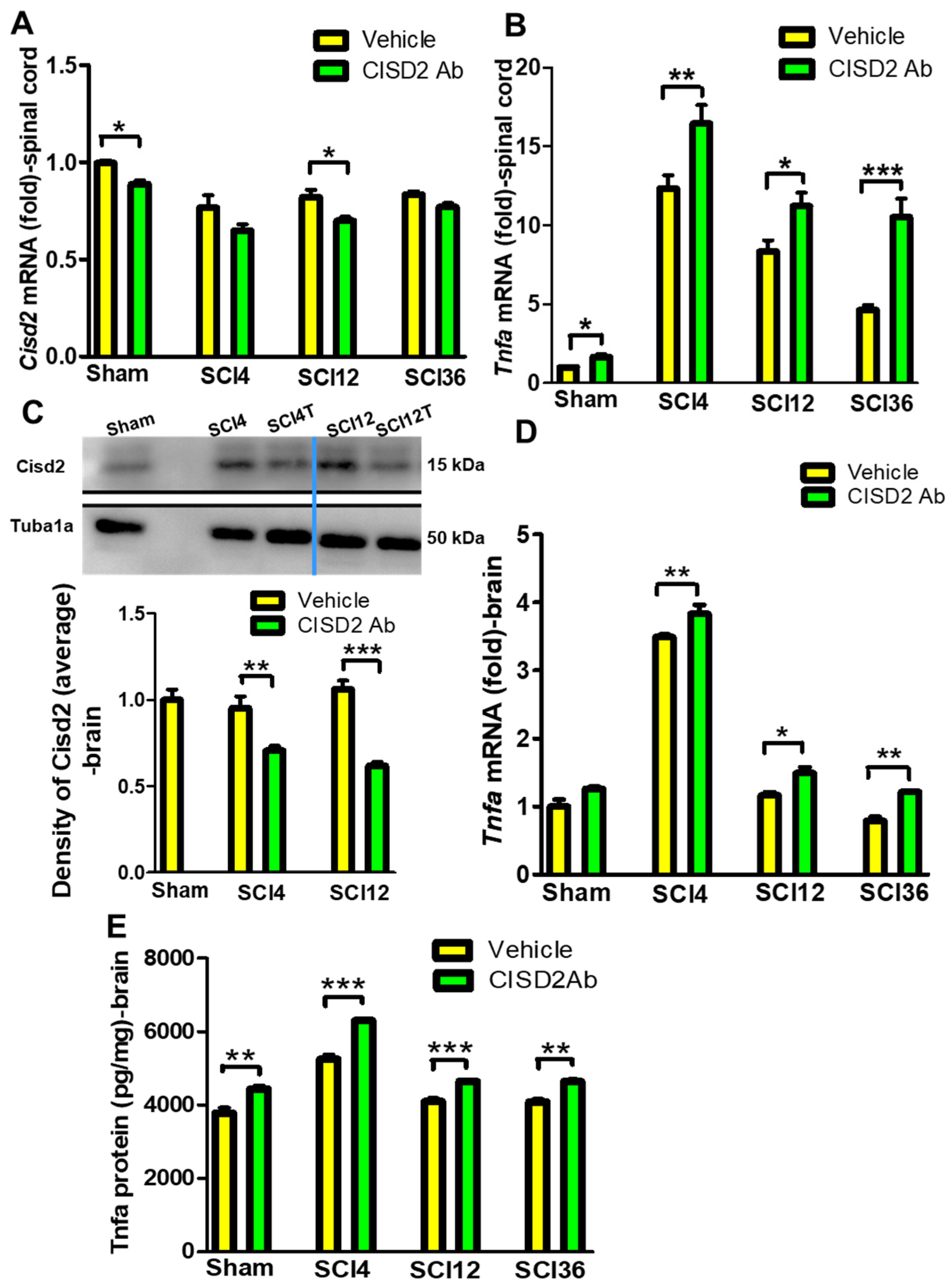
3.3. Inhibition of CISD2 Function Exacerbated Inflammatory Responses in the Brain and Spinal Cord of SCI Mice
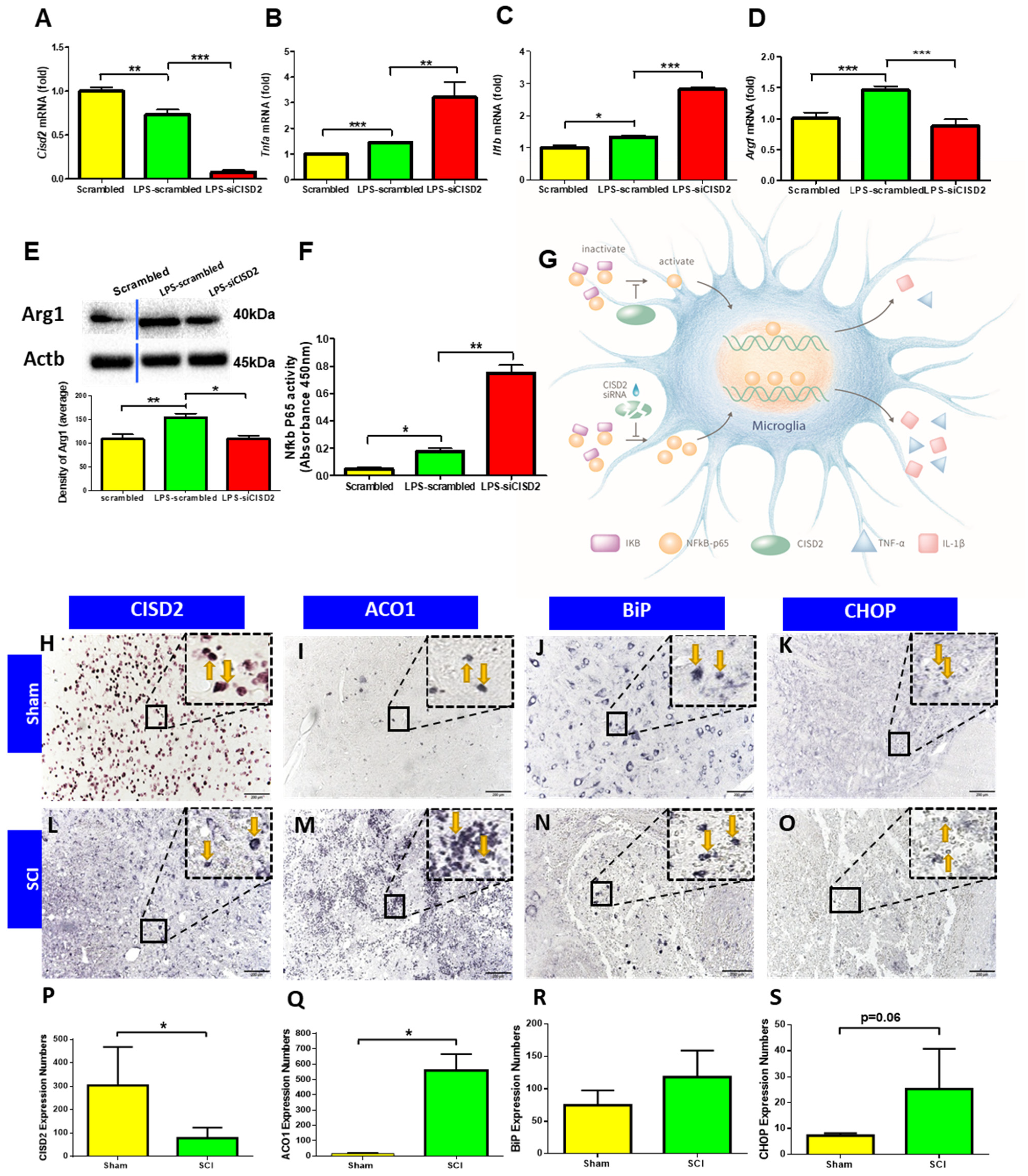
3.4. Cisd2 Suppression Exacerbated LPS-Induced Inflammation in EOC Microglial Cells
3.5. Potentiation of CISD2 Function Using TTM Increased CISD2 Expression and Decreased the Plasma and CSF Levels of Inflammatory Mediators in Patients with CNS Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, C.-C.; Chiang, T.-H.; Sun, Y.-Y.; Lin, M.-S. Protective Effects of CISD2 and Influence of Curcumin on CISD2 Expression in Aged Animals and Inflammatory Cell Model. Nutrients 2019, 11, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, P.; González-Tablas, M.; Otero, Á.; Pascual, D.; Miranda, D.; Ruiz, L.; Sousa, P.; Ciudad, J.; Gonçalves, J.M.; Lopes, M.C.; et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 2016, 53, 1–15. [Google Scholar] [CrossRef]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.-C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.-J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A Homozygous Mutation in a Novel Zinc-Finger Protein, ERIS, Is Responsible for Wolfram Syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; Van Der Geer, P.; Chen, Y.F.; Tsai, T.F.; Simon, M.I.; et al. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol. Med. 2013, 5, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012, 18, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechushtai, R.; Conlan, A.R.; Harir, Y.; Song, L.; Yogev, O.; Eisenberg-Domovich, Y.; Livnah, O.; Michaeli, D.; Rosen, R.; Ma, V.; et al. Characterization of Arabidopsis NEET Reveals an Ancient Role for NEET Proteins in Iron Metabolism. Plant Cell 2012, 24, 2139–2154. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Baur, J.A. A NEET Way to Impair Mitochondrial Function in α- and β-Cells. Diabetes 2016, 65, 1484–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Navarrete, J.M.; Moreno, M.; Ortega, F.J.; Sabater-Masdeu, M.; Xifra, G.; Ricart, W.; Fernández-Real, J.M. CISD1in association with obesity-associated dysfunctional adipogenesis in human visceral adipose tissue. Obesity 2016, 24, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Zhang, L.; Lai, S.; Ye, K. Structure and Molecular Evolution of CDGSH Iron-Sulfur Domains. PLoS ONE 2011, 6, e24790. [Google Scholar] [CrossRef]
- Pesce, L.; Calandrini, V.; Marjault, H.-B.; Lipper, C.H.; Rossetti, G.; Mittler, R.; Jennings, P.A.; Bauer, A.; Nechushtai, R.; Carloni, P. Molecular Dynamics Simulations of the [2Fe–2S] Cluster-Binding Domain of NEET Proteins Reveal Key Molecular Determinants That Induce Their Cluster Transfer/Release. J. Phys. Chem. B 2017, 121, 10648–10656. [Google Scholar] [CrossRef]
- Inupakutika, M.A.; Sengupta, S.; Nechushtai, R.; Jennings, P.A.; Onuchic, J.N.; Azad, R.K.; Padilla, P.; Mittler, R. Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates. Sci. Rep. 2017, 7, srep42571. [Google Scholar] [CrossRef] [PubMed]
- Karmi, O.; Marjault, H.-B.; Pesce, L.; Carloni, P.; Onuchic, J.N.; Jennings, P.A.; Mittler, R.; Nechushtai, R. The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease. JBIC J. Biol. Inorg. Chem. 2018, 23, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.; Lipper, C.H.; Morcos, F.; et al. Structure–function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta 2015, 1853, 1294–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, A.; Krashia, P.; D’Amelio, M. Cisd2: A promising new target in Alzheimer’s disease. J. Pathol. 2020, 251, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Karmi, O.; Holt, S.H.; Song, L.; Tamir, S.; Luo, Y.; Bai, F.; Adenwalla, A.; Darash-Yahana, M.; Sohn, Y.-S.; Jennings, P.A.; et al. Interactions between mitoNEET and NAF-1 in cells. PLoS ONE 2017, 12, e0175796. [Google Scholar] [CrossRef] [Green Version]
- Darash-Yahana, M.; Pozniak, Y.; Lu, M.; Sohn, Y.-S.; Karmi, O.; Tamir, S.; Bai, F.; Song, L.; Jennings, P.A.; Pikarsky, E.; et al. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 10890–10895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlan, A.R.; Axelrod, H.L.; Cohen, A.; Abresch, E.C.; Zuris, J.; Yee, D.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L. Crystal Structure of Miner1: The Redox-active 2Fe-2S Protein Causative in Wolfram Syndrome 2. J. Mol. Biol. 2009, 392, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.C.; Nguyen, M.; Shore, G.C. BCL2-CISD2: An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy 2012, 8, 856–857. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Nguyen, M.; Bourdon, J.; Risse, P.-A.; Martin, J.; Danialou, G.; Rizzuto, R.; Petrof, B.J.; Shore, G.C. Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 2012, 21, 2277–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Kao, C.-H.; Wang, C.-H.; Chen, Y.T.; Wu, C.-Y.; Tsai, C.-Y.; Liu, F.-C.; Yang, C.-W.; Wei, Y.-H.; Hsu, M.-T.; et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, W.-M.; Lin, C.-C.; Kuo, C.-Y.; Juin, Y.-C.; Wu, P.-C.; Lin, M.-S.; Klivényi, P. Wild Bitter Melon Exerts Anti-Inflammatory Effects by Upregulating Injury-Attenuated CISD2 Expression following Spinal Cord Injury. Behav. Neurol. 2020, 2020, 1080521. [Google Scholar] [CrossRef] [PubMed]
- Kung, W.-M.; Chang, C.-J.; Chen, T.-Y.; Lin, M.-S. Cryogen spray cooling mitigates inflammation and injury-induced CISD2 decline in rat spinal cord hemisection model. J. Integr. Neurosci. 2020, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Chiang, T.-H.; Chen, W.-J.; Sun, Y.-Y.; Lee, Y.-H.; Lin, M.-S. CISD2 serves a novel role as a suppressor of nitric oxide signalling and curcumin increases CISD2 expression in spinal cord injuries. Injury 2015, 46, 2341–2350. [Google Scholar] [CrossRef]
- Lin, M.-S. CISD2 Attenuates Inflammation and Regulates Microglia Polarization in EOC Microglial Cells—As a Potential Therapeutic Target for Neurodegenerative Dementia. Front. Aging Neurosci. 2020, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, H.; Schneider, H.; Bösel, J.; Hobohm, C.; Poli, S.; Kollmar, R.; Sobesky, J.; Wolf, S.; Bauer, M.; Tittel, S.; et al. Outcomes of Hypothermia in Addition to Decompressive Hemicraniectomy in Treatment of Malignant Middle Cerebral Artery Stroke: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 571–579. [Google Scholar] [CrossRef]
- Jeong, H.-Y.; Chang, J.-Y.; Yum, K.S.; Hong, J.-H.; Jeong, J.-H.; Yeo, M.-J.; Bae, H.-J.; Han, M.-K.; Lee, K. Extended Use of Hypothermia in Elderly Patients with Malignant Cerebral Edema as an Alternative to Hemicraniectomy. J. Stroke 2016, 18, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.A.; Ko, S.-B.; Presciutti, M.; Fernandez, L.; Carpenter, A.M.; Lesch, C.; Gilmore, E.; Malhotra, R.; Mayer, S.A.; Lee, K.; et al. Prevention of Shivering During Therapeutic Temperature Modulation: The Columbia Anti-Shivering Protocol. Neurocritical Care 2011, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-S.; Lee, Y.-H.; Chiu, W.-T.; Hung, K.-S. Curcumin Provides Neuroprotection After Spinal Cord Injury. J. Surg. Res. 2011, 166, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Pegg, C.C.; He, C.; Stroink, A.R.; Kattner, K.A.; Wang, C.X. Technique for collection of cerebrospinal fluid from the cisterna magna in rat. J. Neurosci. Methods 2010, 187, 8–12. [Google Scholar] [CrossRef]
- Lin, M.-S.; Sun, Y.-Y.; Chiu, W.-T.; Hung, C.-C.; Chang, C.-Y.; Shie, F.-S.; Tsai, S.-H.; Lin, J.-W.; Hung, K.-S.; Lee, Y.-H. Curcumin Attenuates the Expression and Secretion of RANTES after Spinal Cord Injury In Vivo and Lipopolysaccharide-Induced Astrocyte Reactivation In Vitro. J. Neurotrauma 2011, 28, 1259–1269. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y.; et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef]
- Nightingale, T.; Zheng, M.M.Z.; Sachdeva, R.; Phillips, A.A.; Krassioukov, A.V. Diverse cognitive impairment after spinal cord injury is associated with orthostatic hypotension symptom burden. Physiol. Behav. 2020, 213, 112742. [Google Scholar] [CrossRef]
- Sachdeva, R.; Gao, F.; Chan, C.C.H.; Krassioukov, A.V. Cognitive function after spinal cord injury: A systematic review. Neurology 2018, 91, 611–621. [Google Scholar] [CrossRef]
- Craig, A.; Guest, R.; Tran, Y.; Middleton, J. Cognitive Impairment and Mood States after Spinal Cord Injury. J. Neurotrauma 2017, 34, 1156–1163. [Google Scholar] [CrossRef]
- Khazaeipour, Z.; Taheri-Otaghsara, S.-M.; Naghdi, M. Depression Following Spinal Cord Injury: Its Relationship to Demographic and Socioeconomic Indicators. Top. Spinal Cord Inj. Rehab. 2015, 21, 149–155. [Google Scholar] [CrossRef]
- Yeh, T.S.; Ho, Y.C.; Hsu, C.L.; Pan, S.L. Spinal cord injury and Alzheimer’s disease risk: A population-based, retrospective cohort study. Spinal Cord 2018, 56, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.S.; Huang, Y.P.; Wang, H.I.; Pan, S.L. Spinal cord injury and Parkinson’s disease: A population-based, propensity score-matched, longitudinal follow-up study. Spinal Cord 2016, 54, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.-S.; Xie, M.-J.; Yu, Z.-Y.; Zhang, Q.; Wang, Y.-H.; Chen, B.; Chen, C.; Wang, W. Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res. 2007, 1135, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-S.; Chiu, I.-H.; Lin, C.-C. Ultrarapid Inflammation of the Olfactory Bulb After Spinal Cord Injury: Protective Effects of the Granulocyte Colony-Stimulating Factor on Early Neurodegeneration in the Brain. Front. Aging Neurosci. 2021, 13, 701702. [Google Scholar] [CrossRef]
- Altinayar, S.; Oner, S.; Can, S.; Kizilay, A.; Kamisli, S.; Sarac, K. Olfactory disfunction and its relation olfactory bulb volume in Parkinson’s disease. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3659–3664. [Google Scholar]
- Yoo, H.S.; Jeon, S.; Chung, S.J.; Yun, M.; Lee, P.H.; Sohn, Y.H.; Evans, A.C.; Ye, B.S. Olfactory dysfunction in Alzheimer’s disease- and Lewy body-related cognitive impairment. Alzheimers Dement. 2018, 14, 1243–1252. [Google Scholar] [CrossRef]
- Wehling, E.; Naess, H.; Wollschlaeger, D.; Hofstad, H.; Brämerson, A.; Bende, M.; Nordin, S. Olfactory dysfunction in chronic stroke patients. BMC Neurol. 2015, 15, 199. [Google Scholar] [CrossRef] [Green Version]
- Negoias, S.; Hummel, T.; Symmank, A.; Schellong, J.; Joraschky, P.; Croy, I. Olfactory bulb volume predicts therapeutic outcome in major depression disorder. Brain Imaging Behav. 2016, 10, 367–372. [Google Scholar] [CrossRef]
- Croy, I.; Hummel, T. Olfaction as a marker for depression. J. Neurol. 2017, 264, 631–638. [Google Scholar] [CrossRef]
- Yoshizaki, S.; Tamaru, T.; Hara, M.; Kijima, K.; Tanaka, M.; Konno, D.-J.; Matsumoto, Y.; Nakashima, Y.; Okada, S. Microglial inflammation after chronic spinal cord injury is enhanced by reactive astrocytes via the fibronectin/β1 integrin pathway. J. Neuroinflamm. 2021, 18, 12. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Hou, L.; Jing, L.; Ruan, Z.; Peng, B.; Zhang, X.; Hong, J.-S.; Zhao, J.; Wang, Q. Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J. Neuroinflamm. 2021, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Novera, W.; Lee, Z.-W.; Nin, D.S.; Dai, M.Z.-Y.; Idres, S.B.; Wu, H.; Damen, J.M.A.; Tan, T.Z.; Sim, A.Y.L.; Long, Y.C.; et al. Cysteine Deprivation Targets Ovarian Clear Cell Carcinoma Via Oxidative Stress and Iron−Sulfur Cluster Biogenesis Deficit. Antioxid. Redox Signal. 2020, 33, 1191–1208. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truettner, J.S.; Bramlett, H.M.; Dietrich, W.D. Posttraumatic therapeutic hypothermia alters microglial and macrophage polarization toward a beneficial phenotype. J. Cereb. Blood Flow Metab. 2017, 37, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Ahn, J.H.; Cho, J.H.; Tae, H.-J.; Lee, T.-K.; Kim, B.; Lee, J.-C.; Park, J.H.; Shin, M.C.; Ohk, T.G.; et al. Effects of hypothermia on inflammatory cytokine expression in rat liver following asphyxial cardiac arrest. Exp. Ther. Med. 2021, 21, 626. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhang, L.; Liao, J.; Csernus, B.; Kastin, A.J. Selective increase in TNF alpha permeation across the blood-spinal cord barrier after SCI. J. Neuroimmunol. 2003, 134, 111–117. [Google Scholar] [CrossRef]
- Tsai, M.-C.; Wei, C.-P.; Lee, D.-Y.; Tseng, Y.-T.; Shih, Y.-L.; Lee, Y.-H.; Chang, S.-F.; Leu, S.-J. Inflammatory mediators of cerebrospinal fluid from patients with spinal cord injury. Surg. Neurol. 2008, 70, S19–S24. [Google Scholar] [CrossRef]
- Cheng, Y.; Desse, S.; Martinez, A.; Worthen, R.; Jope, R.S.; Beurel, E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav. Immun. 2018, 69, 556–567. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Z.; Sabirzhanov, B.; Stoica, B.A.; Kumar, A.; Luo, T.; Skovira, J.; Faden, A.I. Spinal Cord Injury Causes Brain Inflammation Associated with Cognitive and Affective Changes: Role of Cell Cycle Pathways. J. Neurosci. 2014, 34, 10989–11006. [Google Scholar] [CrossRef]
- Fumagalli, F.; Madaschi, L.; Caffino, L.; Marfia, G.; Di Giulio, A.; Racagni, G.; Gorio, A. Acute spinal cord injury reduces brain derived neurotrohic factor expression in rat hippocampus. Neuroscience 2009, 159, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.K.; Liu, S.; Van Der Poel, C.; McDonald, S.J.; Brady, R.D.; Taylor, L.; Yang, L.; Gardner, A.J.; Ordidge, R.; O’Brien, T.J.; et al. Traumatic Brain Injury Results in Cellular, Structural and Functional Changes Resembling Motor Neuron Disease. Cereb. Cortex 2017, 27, 4503–4515. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Briguglio, F.; Paterniti, I.; Mazzon, E.; Oteri, G.; Militi, D.; Cordasco, G.; Cuzzocrea, S. Emerging role of PPAR-β/δ in inflammatory process associated to experimental periodontitis. Mediators Inflamm. 2011, 2011, 787159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ben Chen, B.; Zhong, X.; Zhou, H.; Zhang, M.; Mai, N.; Wu, Z.; Huang, X.; Haehner, A.; Chen, X.; et al. Olfactory Dysfunction Is Already Present with Subjective Cognitive Decline and Deepens with Disease Severity in the Alzheimer’s Disease Spectrum. J. Alzheimer’s Dis. 2021, 79, 585–595. [Google Scholar] [CrossRef]
- Mesholam, R.I.; Moberg, P.J.; Mahr, R.N.; Doty, R.L. Olfaction in neurodegenerative disease: A meta-analysis of olfactory functioning in Alzheimer's and Parkinson’s diseases. Arch. Neurol. 1998, 55, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Li, Q.; Fang, L.; Cheng, K.; Zhang, R.; Zheng, P.; Zhan, Q.; Qi, Z.; Zhong, S.; Xie, P. Reduced neurogenesis and pre-synaptic dysfunction in the olfactory bulb of a rat model of depression. Neuroscience 2011, 192, 609–618. [Google Scholar] [CrossRef]
- Heeger, P.; Wolf, G.; Meyers, C.; Sun, M.J.; O’Farrell, S.C.; Krensky, A.M.; Neilson, E.G. Isolation and characterization of cDNA from renal tubular epithelium encoding murine Rantes. Kidney Int. 1992, 41, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Doursout, M.F.; Schurdell, M.S.; Young, L.M.; Osuagwu, U.; Hook, D.M.; Poindexter, B.J.; Schiess, M.C.; Bick, D.L.; Bick, R.J. Inflammatory cells and cytokines in the olfactory bulb of a rat model of neuroinflammation; insights into neurodegeneration? J. Interferon Cytokine Res. 2013, 33, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Doty, R.L. Olfaction in Parkinson’s disease and related disorders. Neurobiol. Dis. 2012, 46, 527–552. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.O.; Christianson, T.J.H.; Kremers, W.K.; Mielke, M.; Machulda, M.M.; Vassilaki, M.; Alhurani, R.E.; Geda, Y.E.; Knopman, D.S.; Petersen, R.C. Association Between Olfactory Dysfunction and Amnestic Mild Cognitive Impairment and Alzheimer Disease Dementia. JAMA Neurol. 2016, 73, 93–101. [Google Scholar] [CrossRef]
- Taylor, R. Interpretation of the Correlation Coefficient: A Basic Review. J. Diagn. Med. Sonogr. 1990, 6, 35–39. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chou, T.; Lin, I.H.; Chen, C.G.; Kao, C.H.; Huang, G.J.; Chen, L.K.; Wang, P.N.; Lin, C.P.; Tsai, T.F. Upregulation of Cisd2 attenuates Alzheimer’s-related neuronal loss in mice. J. Pathol. 2020, 250, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, J.A.; Massé, E. The iron-sensing aconitase B binds its own mRNA to prevent sRNA-induced mRNA cleavage. Nucleic Acids Res. 2014, 42, 10023–10036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age (Years) | Sex | Etiology of CNS Insults | Cause of Injury | Hematoma Location | GCS Score at the Time of Admission | Surgery | TTM Treatment | Plasma CISD2 Levels (ng/mL) | CSF CISD2 Levels (ng/mL) | Plasma IL6 Levels (ng/mL) | CSF IL6 Levels (ng/mL) | Plasma CRP Levels (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 77 | F | Hemorrhagic stroke | - | ICH, right BG | E1V1M1 | CN+HR+ EVD | - | 2.04 | 0.25 | 133.28 | 1379.27 | - |
| 2 | 54 | M | Hemorrhagic stroke | - | ICH, right BG | E1V1M4 | EVD | + | 3.13 | 0.30 | 69.57 | 1477.40 | 18.68 |
| 2 post-TTM | 4.89 | 0.63 | 559.69 | 1160.13 | 7.43 | ||||||||
| 3 | 53 | F | Hemorrhagic stroke | - | Cerebellum | E2V1M5 | EVD | - | 2.45 | - | 15.82 | - | - |
| 4 | 16 | M | TBI | Motor vehicle accident | Acute EDH,SDH, right F-T | E2V1M5 | Uni + HR + EVD | - | 2.09 | 0.45 | 77.70 | 242.59 | - |
| 5 | 53 | M | TBI | Fall | Acute SDH, right F-T | E1V1M1 | Uni + HR + EVD | - | 1.36 | 0.29 | 177.98 | 1374.02 | - |
| 6 | 18 | F | TBI | Motor vehicle accident | Acute SDH, right F-T | E1V1M1 | Uni + HR + EVD | - | 0.46 | - | 1312.53 | - | - |
| 7 | 44 | M | Parasagittal meningioma | - | - | E4V5M6 | Removal of tumor | - | 3.21 | - | 9.53 | - | - |
| 8 | 20 | F | TBI | Motor vehicle accident | Acute SDH, right F-T | E4V5M6 | - | - | 3.68 | - | 22.78 | - | - |
| 9 | 57 | M | Hemorrhagic stroke | - | ICH, left BG | E3V1M5 | EVD | - | 2.41 | 0.62 | 9.86 | 624.83 | - |
| 10 | 23 | M | Burst fracture of the first lumbar vertebra with spinal EDH, SCI (AIS-B) | Fall | Spinal EDH | E4V5M6 | HR+ fusion procedure | - | 3.30 | - | 41.79 | - | - |
| 11 | 66 | M | Hemorrhagic stroke | - | ICH, left BG | E1V1M1 | Uni + HR + EVD | - | 3.12 | 0.24 | 81.08 | 1367.49 | - |
| 12 | 21 | M | Burst fracture of the fifth and sixth cervical vertebra, SCI (AIS-A) | Motor vehicle accident | - | E4V5M6 | Cervical corpectomy + fusion procedure + lumbar drain | + | 1.78 | 0.39 | 3.51 | 1254.79 | 1.06 |
| 12 post-TTM | 2.38 | 0.54 | 27.64 | 284.05 | 0.46 | ||||||||
| 13 | 57 | M | TBI | Motor vehicle accident | Acute SDH, right F-T-P | E1V1M3 | Uni + HR + EVD | + | 2.85 | 0.34 | 105.32 | 867.87 | 3.28 |
| 13 post-TTM | 3.12 | 0.37 | 231.25 | 112.38 | 1.30 | ||||||||
| Control | |||||||||||||
| 1 | 45 | M | 3.60 | - | 0.00 | - | - | ||||||
| 2 | 39 | M | 4.33 | - | 0.21 | - | - | ||||||
| 3 | 37 | M | 3.12 | - | 1.63 | - | - |
| Mice | ||
|---|---|---|
| Gene | Orientation | Sequence |
| Cisd2 | forward | 5′-AAAGCAGCTTATTGTAGGTGCTG-3′ |
| reverse | 5′-CGCCTGTCAATTCATTATGC-3′ | |
| Tnfa | forward | 5′-GTGAAGGGAATGGGTGTT-3′ |
| reverse | 5′-GGTCACTGTCCCAGCATC-3′ | |
| Actb | forward | 5′-CACTGTGCCCATCTACGA-3′ |
| reverse | 5′-ATGTCACGCACGATTTCC-3′ | |
| EOC microglial cell | ||
| Cisd2 | forward | 5′-AAAATCCCAAGGTGGTGAATGA-3′ |
| reverse | 5′-GGACCGCCAGCACCTACA-3′ | |
| Tnfa | forward | 5′-GGCACTCCCCCAAAAGATG-3′ |
| reverse | 5′-GCCACAAGCAGGAATGAGA-3′ | |
| Il1b | forward | 5′-AAGATGAAGGGCTGCTTCCA-3′ |
| reverse | 5′-ATGTGCTGCTGCGAGATTTG-3′ | |
| Arg1 | forward | 5′-CGCCTTTCTCAAAAGGACAG-3′ |
| reverse | 5′-CCAGCTCTTCATTGGCTTTC-3′ | |
| Gapdh | forward | 5′-GGCAAATTCAACGGCACAGT-3′ |
| reverse | 5′-CGCTCCTGGAAGATGGTGAT-3′ | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kung, W.-M.; Lin, C.-C.; Chen, W.-J.; Jiang, L.-L.; Sun, Y.-Y.; Hsieh, K.-H.; Lin, M.-S. Anti-Inflammatory CDGSH Iron-Sulfur Domain 2: A Biomarker of Central Nervous System Insult in Cellular, Animal Models and Patients. Biomedicines 2022, 10, 777. https://doi.org/10.3390/biomedicines10040777
Kung W-M, Lin C-C, Chen W-J, Jiang L-L, Sun Y-Y, Hsieh K-H, Lin M-S. Anti-Inflammatory CDGSH Iron-Sulfur Domain 2: A Biomarker of Central Nervous System Insult in Cellular, Animal Models and Patients. Biomedicines. 2022; 10(4):777. https://doi.org/10.3390/biomedicines10040777
Chicago/Turabian StyleKung, Woon-Man, Chai-Ching Lin, Wei-Jung Chen, Li-Lin Jiang, Yu-Yo Sun, Kuang-Hui Hsieh, and Muh-Shi Lin. 2022. "Anti-Inflammatory CDGSH Iron-Sulfur Domain 2: A Biomarker of Central Nervous System Insult in Cellular, Animal Models and Patients" Biomedicines 10, no. 4: 777. https://doi.org/10.3390/biomedicines10040777