
Kinetic Processes in Enzymatic Nanoreactors for In Vivo Detoxification



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Principles of Detoxification Processes by Exogenous Enzymes: Stoichiometric Neutralization versus Catalytic and Pseudo-Catalytic Degradation of Toxicants
2.1. In Vivo Detoxification by Free Enzymes
2.1.1. Catalytic Neutralization
2.1.2. Stoichiometric Neutralization
2.1.3. Pseudo-Catalytic Neutralization
2.2. Detoxification by Enzymes Encapsulated in Circulating Nanoreactors
2.2.1. Structure of Enzyme-Containing Nanoreactors and their Application to Improve Biocatalytic Reactions
- -
- Increasing enzyme-loading into the nanoreactor [30]: This can be achieved by: (i) the modification of enzymes for strengthening their binding in nanoreactor, which can be carried out by covalent bonds with small molecules or polymers, electrostatic bonds and their immobilization on polymer, and by using cross-linking agents. One of the most successful examples is of three enzymes covalently encapsulated into a substrate-permeable silica nanoreactor by a mild fluoride-catalyzed sol–gel process [29]. (ii) The multicompartment loading of enzymes and the sub-compartmentalization of several enzymes by encapsulation within the lumen, entrapment in the membrane, and conjugation on the surface may yield high efficiency. This can be achieved by the cross-linking of enzymes with the building blocks of nanoreactors, or by capturing them in a layer-by-layer assembly. Incompatible enzymes can be encapsulated in nanoreactor sub-compartments. In addition, the mutual templated crystallization between enzymes and the material of the nanoreactor prevents enzyme molecules from unfolding [31]. Recently, a dual-confinement strategy that can controllably confine a series of enzymes in a nanocage-based zeolite imidazole framework [32] and multi-shelled metal-organic frameworks [33] were developed. (iii) The control of nanoreactor self-assembly by the regulation of parameters—packing parameter (p) and polymer hydrophilicity (ƒw)—and using improved innovative methods for nanoreactor biotechnology. Thus, microfluidics presents a new and promising method, providing high encapsulation efficiency and scaling [34].
- -
- The permeability to reactants and reaction products needs to be improved. This can be achieved through different strategies: (i) the selective selection of nanoreactor building blocks. Several methods of quantifying membrane permeability (fluorescence spectroscopy, osmotic swelling, pulsed-field gradient NMR spectroscopy) and the passage of molecules have been proposed [35]. Usually, lipid vesicles are highly permeable. Further strategies are associated with the production of organo-inorganic hybrid and polymeric vesicles. Polymeric vesicles are generally impermeable, due to their increased thickness and low membrane lateral diffusion. Diffusion is primarily associated with different structural conformations of the macromolecules in the membrane. Thus, hindered diffusion was observed for copolymers of high molecular weight [36]. The permeability of hybrid unilamellar polymer/lipid vesicles is mainly determined by the lipid-polymer composition [37]. The influence of the molecular weight and architecture of block copolymers on the linear tension and its consequences for the membrane structuring of hybrid polymer-lipid vesicles are presented in the literature [38]. One innovative strategy is switching between the impermeable and a semipermeable state of the vesicle membrane. For this purpose, functional groups were introduced into the block copolymer membrane for increasing its polarity in response to mechanical action, thereby increasing the permeability of the membrane to water-soluble substrates [39]. Another new approach is the incorporation of the polymer into a phospholipid membrane, where it acts as a synthetic molecular channel. Imparting an anionic charge to the vesicle’s surface represents a simple and versatile approach to substrate sorting and enhances molecular permeability [40]. The unique lattice membrane of the vesicles was made up of hundreds of small polymeric vesicles, linked together through multiple interactions [41]. (ii) The application of stimulus-responsive materials for the synthesis of nanoreactors, such as temperature, light, solvents, pH, and light [18]. Such changes often alter permeability or the phase separation dynamics; thus, they can be used to effectively turn on or off enzymatic reactions. Examples of photo-cross-linked and pH-sensitive materials for polymersomes are presented in the literature [42]. The bidirectional feedback mechanism of chemoenzymatic pH clock-mediated transient assembly regulates the existence of the transient state and controls the activity of the assembly [43]. Stimulus-responsive linkers were integrated into the cross-linking membrane network of nanoreactors, based on polyion complex vesicles [44]. The permeability of polymer-functionalized epoxy membranes was modulated by opening the epoxy ring and various diamine crosslinkers and hydrophobic primary amines [45]. (iii) The insertion of membrane proteins and the formation of membrane protein channels [46] via peptides that form bio-pores. Thus, the design of “intelligent” membranes with precisely controlled and sensitive behavior may provide solutions to increase the efficiency of nanoreactors.
2.2.2. Concentration of Enzymes inside the Nanoreactor
2.2.3. Kinetic Principles of Enzyme Reactions in Nanoreactors
2.2.4. Viscosity Effects
2.2.5. Osmotic Effects
2.2.6. Macromolecular Crowding
2.3. Practical Implication of Second-Order Reactions in Nanoreactors
2.3.1. General Considerations
2.3.2. Stoichiometric and Pseudo-Catalytic Detoxification under Second-Order Processes
2.3.3. Catalytic Detoxification
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oerlemans, R.A.J.F.; Timmermans, S.B.P.E.; Hest, J.C.M. Artificial Organelles: Towards Adding or Restoring Intracellular Activity. ChemBioChem 2021, 22, 2051–2078. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, K.; Zárate-Romero, A.; Sengar, P.; Medrano, C.; Vazquez-Duhalt, R. Catalytic Kinetics Considerations and Molecular Tools for the Design of Multienzymatic Cascade Nanoreactors. ChemCatChem 2021, 13, 3732–3748. [Google Scholar] [CrossRef]
- Godoy-Gallardo, M.; York-Duran, M.J.; Hosta-Rigau, L. Recent Progress in Micro/Nanoreactors toward the Creation of Artificial Organelles. Adv. Healthc. Mater. 2018, 7, 170091. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Zhu, T.; Li, J.; Ye, Z.; Shi, C.; Guo, Z.; Wang, J.; Chen, X.; Zheng, N. A Novel Cascade Nanoreactor Integrating Two-Dimensional Pd-Ru Nanozyme, Uricase and Red Blood Cell Membrane for Highly Efficient Hyperuricemia Treatment. Small 2021, 17, 2103645. [Google Scholar] [CrossRef]
- Forster, V.; Leroux, J.-C. Nano-antidotes for drug overdose and poisoning. Sci. Transl. Med. 2015, 7, 290ps14. [Google Scholar] [CrossRef]
- Szilasi, M.; Budai, M.; Budai, L.; Petrikovics, I. Nanoencapsulated and microencapsulated enzymes in drug antidotal therapy. Toxicol. Ind. Health 2012, 28, 522–531. [Google Scholar] [CrossRef]
- Pashirova, T.N.; Bogdanov, A.; Masson, P. Therapeutic nanoreactors for detoxification of xenobiotics: Concepts, challenges and biotechnological trends with special emphasis to organophosphate bioscavenging. Chem. Biol. Interact. 2021, 346, 109577. [Google Scholar] [CrossRef]
- Eyer, F.; Eyer, P. Enzyme-based assay for quantification of paraoxon in blood of parathion poisoned patients. Hum. Exp. Toxicol. 1998, 17, 645–651. [Google Scholar] [CrossRef]
- Küchler, A.; Yoshimoto, M.; Luginbühl, S.; Mavelli, F.; Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409–420. [Google Scholar] [CrossRef]
- Worek, F.; Thiermann, H.; Szinicz, L.; Eyer, P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem. Pharmacol. 2004, 68, 2237–2248. [Google Scholar] [CrossRef]
- Zorbaz, T.; Braïki, A.; Maraković, N.; Renou, J.; de la Mora, E.; Maček Hrvat, N.; Katalinić, M.; Silman, I.; Sussman, J.L.; Mercey, G.; et al. Potent 3-Hydroxy-2-Pyridine Aldoxime Reactivators of Organophosphate-Inhibited Cholinesterases with Predicted Blood–Brain Barrier Penetration. Chem.-A Eur. J. 2018, 24, 9675–9691. [Google Scholar] [CrossRef] [PubMed]
- Maček Hrvat, N.; Kalisiak, J.; Šinko, G.; Radić, Z.; Sharpless, K.B.; Taylor, P.; Kovarik, Z. Evaluation of high-affinity phenyltetrahydroisoquinoline aldoximes, linked through anti-triazoles, as reactivators of phosphylated cholinesterases. Toxicol. Lett. 2020, 321, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Nachon, F.; Lockridge, O. Structural approach to the aging of phosphylated cholinesterases. Chem. Biol. Interact. 2010, 187, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, Z.; Maček Hrvat, N. Efficient detoxification of nerve agents by oxime-assisted reactivation of acetylcholinesterase mutants. Neuropharmacology 2020, 171, 108111. [Google Scholar] [CrossRef]
- Pang, Z.; Hu, C.M.J.; Fang, R.H.; Luk, B.T.; Gao, W.; Wang, F.; Chuluun, E.; Angsantikul, P.; Thamphiwatana, S.; Lu, W.; et al. Detoxification of Organophosphate Poisoning Using Nanoparticle Bioscavengers. ACS Nano 2015, 9, 6450–6458. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, E.J.; Tsao, C.; Kasten, S.A.; Boeri, M.V.; Dao, T.L.; DeBus, S.J.; Cadieux, C.L.; Baker, C.A.; Otto, T.C.; et al. Nanoscavenger provides long-term prophylactic protection against nerve agents in rodents. Sci. Transl. Med. 2019, 11, eaau7091. [Google Scholar] [CrossRef] [Green Version]
- Einfalt, T.; Garni, M.; Witzigmann, D.; Sieber, S.; Baltisberger, N.; Huwyler, J.; Meier, W.; Palivan, C.G. Bioinspired Molecular Factories with Architecture and In Vivo Functionalities as Cell Mimics. Adv. Sci. 2020, 7, 1901923. [Google Scholar] [CrossRef]
- Araste, F.; Aliabadi, A.; Abnous, K.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Self-assembled polymeric vesicles: Focus on polymersomes in cancer treatment. J. Control. Release 2021, 330, 502–528. [Google Scholar] [CrossRef]
- Ke, W.; Li, J.; Mohammed, F.; Wang, Y.; Tou, K.; Liu, X.; Wen, P.; Kinoh, H.; Anraku, Y.; Chen, H.; et al. Therapeutic polymersome nanoreactors with tumor-specific activable cascade reactions for cooperative cancer therapy. ACS Nano 2019, 13, 2357–2369. [Google Scholar] [CrossRef]
- Sueyoshi, D.; Anraku, Y.; Komatsu, T.; Urano, Y.; Kataoka, K. Enzyme-Loaded Polyion Complex Vesicles as in Vivo Nanoreactors Working Sustainably under the Blood Circulation: Characterization and Functional Evaluation. Biomacromolecules 2017, 18, 1189–1196. [Google Scholar] [CrossRef]
- Li, X.; Zhao, X.; Lv, R.; Hao, L.; Huo, F.; Yao, X. Polymeric Nanoreactors as Emerging Nanoplatforms for Cancer Precise Nanomedicine. Macromol. Biosci. 2021, 21, 2000424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Lu, Y. Enzyme therapeutics for systemic detoxification. Adv. Drug Deliv. Rev. 2015, 90, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Qi, G.; Ma, K.; Qu, X.; Xu, W.; Xu, S.; Jin, Y. Tumor Microenvironment-Activated Degradable Multifunctional Nanoreactor for Synergistic Cancer Therapy and Glucose SERS Feedback. iScience 2020, 23, 101274. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Jia, X.; Zhong, L.; Jiao, Y.; Zhang, Z.; Wang, Z.; Feng, Y.; Bilal, M.; Cui, J.; Jia, S. Metal-organic frameworks with different dimensionalities: An ideal host platform for enzyme@MOF composites. Coord. Chem. Rev. 2022, 454, 214327. [Google Scholar] [CrossRef]
- Chen, C.; Vázquez-González, M.; O’Hagan, M.P.; Ouyang, Y.; Wang, Z.; Willner, I. Enzyme-Loaded Hemin/G-Quadruplex-Modified ZIF-90 Metal–Organic Framework Nanoparticles: Bioreactor Nanozymes for the Cascaded Oxidation of N -hydroxy- l -arginine and Sensing Applications. Small 2022, 18, 2104420. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, B.; Zhao, Y.; Fu, J.; Wen, T.; Liao, K.; Quan, G.; Pan, X.; Wu, C. Multifunctional nanoreactors-integrated microneedles for cascade reaction-enhanced cancer therapy. J. Control. Release 2021, 339, 335–349. [Google Scholar] [CrossRef]
- Hang, L.; Zhang, T.; Wen, H.; Li, M.; Liang, L.; Tang, X.; Zhou, C.; Tian, J.; Ma, X.; Jiang, G. Rational design of non-toxic GOx-based biocatalytic nanoreactor for multimodal synergistic therapy and tumor metastasis suppression. Theranostics 2021, 11, 10001. [Google Scholar] [CrossRef]
- Liu, Y.; Du, J.; Yan, M.; Lau, M.Y.; Hu, J.; Han, H.; Yang, O.O.; Liang, S.; Wei, W.; Wang, H.; et al. Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication. Nat. Nanotechnol. 2013, 8, 187–192. [Google Scholar] [CrossRef]
- Jo, S.; Wurm, F.R.; Landfester, K. Enzyme-Loaded Nanoreactors Enable the Continuous Regeneration of Nicotinamide Adenine Dinucleotide in Artificial Metabolisms. Angew. Chem. Int. Ed. 2021, 60, 7728–7734. [Google Scholar] [CrossRef]
- Gaur, D.; Dubey, N.C.; Tripathi, B.P. Biocatalytic self-assembled synthetic vesicles and coacervates: From single compartment to artificial cells. Adv. Colloid Interface Sci. 2022, 299, 102566. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, F.; Lin, Z.; Hu, J.; Shen, R.; Lin, Y.; Liu, X.Y. Silk Nanococoons: Bio-Nanoreactors for Enzymatic Catalytic Reactions and Applications to Alcohol Intoxication. Small Sci. 2021, 1, 2000049. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, M.; Xiong, C.; Zhu, X.; Chen, C.; Zhou, F.; Dong, Y.; Wang, Y.; Xu, J.; Li, Y.; et al. Dual confinement of high–loading enzymes within metal–organic frameworks for glucose sensor with enhanced cascade biocatalysis. Biosens. Bioelectron. 2022, 196, 113695. [Google Scholar] [CrossRef] [PubMed]
- Man, T.; Xu, C.; Liu, X.-Y.; Li, D.; Tsung, C.-K.; Pei, H.; Wan, Y.; Li, L. Hierarchically encapsulating enzymes with multi-shelled metal-organic frameworks for tandem biocatalytic reactions. Nat. Commun. 2022, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Li, S.; Li, X.; Zhan, Q.; Li, L.; Long, L.; Zhao, J.; Hou, X.; Yuan, X. Construction of enzymatic nanoreactors with high catalytic activity in millifluidic systems for cancer therapy. Chem. Eng. J. 2022, 429, 132305. [Google Scholar] [CrossRef]
- Miller, A.J.; Pearce, A.K.; Foster, J.C.; O’Reilly, R.K. Probing and Tuning the Permeability of Polymersomes. ACS Cent. Sci. 2021, 7, 30–38. [Google Scholar] [CrossRef]
- Itel, F.; Chami, M.; Najer, A.; Lörcher, S.; Wu, D.; Dinu, I.A.; Meier, W. Molecular Organization and Dynamics in Polymersome Membranes: A Lateral Diffusion Study. Macromolecules 2014, 47, 7588–7596. [Google Scholar] [CrossRef]
- Fauquignon, M.; Courtecuisse, E.; Josselin, R.; Mutschler, A.; Brûlet, A.; Schmutz, M.; Le Meins, J.-F. Large hybrid Polymer/Lipid Unilamellar vesicle (LHUV) at the nanoscale: An insight into the lipid distribution in the membrane and permeability control. J. Colloid Interface Sci. 2021, 604, 575–583. [Google Scholar] [CrossRef]
- Fauquignon, M.; Ibarboure, E.; Le Meins, J.-F. Hybrid polymer/lipid vesicles: Influence of polymer architecture and molar mass on line tension. Biophys. J. 2022, 121, 61–67. [Google Scholar] [CrossRef]
- Rifaie-Graham, O.; Galensowske, N.F.B.; Dean, C.; Pollard, J.; Balog, S.; Gouveia, M.G.; Chami, M.; Vian, A.; Amstad, E.; Lattuada, M.; et al. Shear Stress-Responsive Polymersome Nanoreactors Inspired by the Marine Bioluminescence of Dinoflagellates. Angew. Chem.-Int. Ed. 2021, 60, 904–909. [Google Scholar] [CrossRef]
- Nishimura, T.; Hirose, S.; Sasaki, Y.; Akiyoshi, K. Substrate-Sorting Nanoreactors Based on Permeable Peptide Polymer Vesicles and Hybrid Liposomes with Synthetic Macromolecular Channels. J. Am. Chem. Soc. 2020, 142, 154–161. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Q.; Wang, F.; Sun, H.; Xiao, J.; Cornel, E.J.; Zhu, Y.; Du, J. Giant Polymer Vesicles with a Latticelike Membrane. ACS Macro Lett. 2021, 10, 1015–1022. [Google Scholar] [CrossRef]
- Moreno, S.; Voit, B.; Gaitzsch, J. The chemistry of cross-linked polymeric vesicles and their functionalization towards biocatalytic nanoreactors. Colloid Polym. Sci. 2020, 299, 309–324. [Google Scholar] [CrossRef]
- Das, S.; Das, P.; Dowari, P.; Kanti Das, B.; Das, D. Bi-Directional Feedback Controlled Transience in Cucurbituril Based Tandem Nanozyme. J. Colloid Interface Sci. 2022, 614, 172–180. [Google Scholar] [CrossRef]
- Li, J.; Anraku, Y.; Kataoka, K. Self-Boosting Catalytic Nanoreactors Integrated with Triggerable Crosslinking Membrane Networks for Initiation of Immunogenic Cell Death by Pyroptosis. Angew. Chem.-Int. Ed. 2020, 59, 13526–13530. [Google Scholar] [CrossRef] [PubMed]
- Varlas, S.; Foster, J.C.; Georgiou, P.G.; Keogh, R.; Husband, J.T.; Williams, D.S.; O’Reilly, R.K. Tuning the membrane permeability of polymersome nanoreactors developed by aqueous emulsion polymerization-induced self-assembly. Nanoscale 2019, 11, 12643–12654. [Google Scholar] [CrossRef] [Green Version]
- Marušič, N.; Otrin, L.; Zhao, Z.; Lira, R.B.; Kyrilis, F.L.; Hamdi, F.; Kastritis, P.L.; Vidaković-Koch, T.; Ivanov, I.; Sundmacher, K.; et al. Constructing artificial respiratory chain in polymer compartments: Insights into the interplay between bo 3 oxidase and the membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 15006–15017. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, M.R.; Mazaheri, E.; Dormiani, K. Simple Equations Pertaining to the Particle Number and Surface Area of Metallic, Polymeric, Lipidic and Vesicular Nanocarriers. Sci. Pharm. 2021, 89, 15. [Google Scholar] [CrossRef]
- Vogel, R.; Savage, J.; Muzard, J.; Della Camera, G.; Vella, G.; Law, A.; Marchioni, M.; Mehn, D.; Geiss, O.; Peacock, B.; et al. Measuring particle concentration of multimodal synthetic reference materials and extracellular vesicles with orthogonal techniques: Who is up to the challenge? J. Extracell. Vesicles 2021, 10, e12052. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Pallares, R.M.; Thanh, N.T.K. Characterization techniques for nanoparticles: Comparison and complementarity upon studying nanoparticle properties. Nanoscale 2018, 10, 12871–12934. [Google Scholar] [CrossRef] [Green Version]
- Minelli, C.; Bartczak, D.; Peters, R.; Rissler, J.; Undas, A.; Sikora, A.; Sjöström, E.; Goenaga-Infante, H.; Shard, A.G. Sticky Measurement Problem: Number Concentration of Agglomerated Nanoparticles. Langmuir 2019, 35, 4927–4935. [Google Scholar] [CrossRef]
- Baalousha, M.; Prasad, A.; Lead, J.R. Quantitative measurement of the nanoparticle size and number concentration from liquid suspensions by atomic force microscopy. Environ. Sci. Process. Impacts 2014, 16, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Khlebtsov, B.N.; Khanadeev, V.A.; Khlebtsov, N.G. Determination of the Size, Concentration, and Refractive Index of Silica Nanoparticles from Turbidity Spectra. Langmuir 2008, 24, 8964–8970. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.-Y.; Tang, M.; Hu, J.; Wu, L.-L.; Pang, D.-W.; Zeng, J.-B.; Li, X.-Y. Determination of the Absolute Number Concentration of Nanoparticles and the Active Affinity Sites on Their Surfaces. Anal. Chem. 2016, 88, 10134–10142. [Google Scholar] [CrossRef] [PubMed]
- Vaclavek, T.; Prikryl, J.; Foret, F. Resistive pulse sensing as particle counting and sizing method in microfluidic systems: Designs and applications review. J. Sep. Sci. 2019, 42, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, J.; Minelli, C.; Hamilton, D.; Wywijas, M.; Jones, H.J. Nanoparticle number concentration measurements by multi-angle dynamic light scattering. J. Nanopart. Res. 2020, 22, 108. [Google Scholar] [CrossRef]
- Pauw, B.R.; Kästner, C.; Thünemann, A.F. Nanoparticle size distribution quantification: Results of a small-angle X-ray scattering inter-laboratory comparison. J. Appl. Crystallogr. 2017, 50, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Clement, S.; Gardner, B.; Razali, W.A.W.; Coleman, V.A.; Jämting, Å.K.; Catchpoole, H.J.; Goldys, E.M.; Herrmann, J.; Zvyagin, A. Quantification of nanoparticle concentration in colloidal suspensions by a non-destructive optical method. Nanotechnology 2017, 28, 475702. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Guha, S.; Zangmeister, R.; Tarlov, M.J.; Zachariah, M.R. Method for Determining the Absolute Number Concentration of Nanoparticles from Electrospray Sources. Langmuir 2011, 27, 14732–14739. [Google Scholar] [CrossRef]
- Epstein, H.; Afergan, E.; Moise, T.; Richter, Y.; Rudich, Y.; Golomb, G. Number-concentration of nanoparticles in liposomal and polymeric multiparticulate preparations: Empirical and calculation methods. Biomaterials 2006, 27, 651–659. [Google Scholar] [CrossRef]
- Urey, C.; Weiss, V.U.; Gondikas, A.; von der Kammer, F.; Hofmann, T.; Marchetti-Deschmann, M.; Allmaier, G.; Marko-Varga, G.; Andersson, R. Combining gas-phase electrophoretic mobility molecular analysis (GEMMA), light scattering, field flow fractionation and cryo electron microscopy in a multidimensional approach to characterize liposomal carrier vesicles. Int. J. Pharm. 2016, 513, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Shard, A.G.; Wright, L.; Minelli, C. Robust and accurate measurements of gold nanoparticle concentrations using UV-visible spectrophotometry. Biointerphases 2018, 13, 061002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Cacioppo, M.; Megahed, S.; Arcudi, F.; Đorđević, L.; Zhu, D.; Schulz, F.; Prato, M.; Parak, W.J.; Feliu, N. Influence of the chirality of carbon nanodots on their interaction with proteins and cells. Nat. Commun. 2021, 12, 7208. [Google Scholar] [CrossRef] [PubMed]
- Tuoriniemi, J.; Moreira, B.; Safina, G. Determining Number Concentrations and Diameters of Polystyrene Particles by Measuring the Effective Refractive Index of Colloids Using Surface Plasmon Resonance. Langmuir 2016, 32, 10632–10640. [Google Scholar] [CrossRef]
- Cuello-Nuñez, S.; Abad-Álvaro, I.; Bartczak, D.; del Castillo Busto, M.E.; Ramsay, D.A.; Pellegrino, F.; Goenaga-Infante, H. The accurate determination of number concentration of inorganic nanoparticles using spICP-MS with the dynamic mass flow approach. J. Anal. At. Spectrom. 2020, 35, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
- Malloy, A.; Carr, B. NanoParticle Tracking Analysis—The HaloTM System. Part. Part. Syst. Charact. 2006, 23, 197–204. [Google Scholar] [CrossRef]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Usfoor, Z.; Kaufmann, K.; Rakib, A.S.H.; Hergenröder, R.; Shpacovitch, V. Features of Sizing and Enumeration of Silica and Polystyrene Nanoparticles by Nanoparticle Tracking Analysis (NTA). Sensors 2020, 20, 6611. [Google Scholar] [CrossRef]
- Laidler, K.J.; Bunting, P.S. [9] The kinetics of immobilized enzyme systems. In Methods in Enzymology; Academic Press/Elsevier: Cambridge, MA, USA, 1980; pp. 227–248. [Google Scholar]
- Dalziel, K. The Chemical Kinetics of Enzyme Reactions. Biochem. Educ. 1974, 2, 32. [Google Scholar] [CrossRef]
- Sundaram, P.V.; Tweedale, A.; Laidler, K.J. Kinetic laws for solid-supported enzymes. Can. J. Chem. 1970, 48, 1498–1504. [Google Scholar] [CrossRef]
- Cha, S. Kinetic Behavior at High Enzyme Concentrations. J. Biol. Chem. 1970, 245, 4814–4818. [Google Scholar] [CrossRef]
- Halfman, C.J.; Marcus, F. A method for determining kinetic parameters at high enzyme concentrations. Biochem. J. 1982, 203, 339–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, S.; Maini, P.K. A century of enzyme kinetics: Reliability of the KM and vmax estimates. Comments Theor. Biol. 2003, 8, 169–187. [Google Scholar] [CrossRef]
- Schnell, S.; Maini, P.K. Enzyme kinetics at high enzyme concentration. Bull. Math. Biol. 2000, 62, 483–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzafriri, A.R. Michaelis-Menten Kinetics at High Enzyme Concentrations. Bull. Math. Biol. 2003, 65, 1111–1129. [Google Scholar] [CrossRef]
- Nakatani, H.; Dunford, H.B. Meaning of diffusion-controlled association rate constants in enzymology. J. Phys. Chem. 1979, 83, 2662–2665. [Google Scholar] [CrossRef]
- Brouwer, A.C.; Kirsch, J.F. Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry 1982, 21, 1302–1307. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Dreher, R.; Davey, J.P. The association reaction of yeast alcohol dehydrogenase with coenzyme is partly diffusion-controlled in solvents of increased viscosity. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1987, 911, 53–58. [Google Scholar] [CrossRef]
- Eigen, M.; Hammes, G.G. Elementary Steps in Enzyme Reactions (as Studied by Relaxation Spectrometry). In Adcances in Enzymology and Related Areas of Molecular Biology; Wiley: New York, NY, USA, 2006; Volome 25, pp. 1–38. [Google Scholar]
- Parsegian, V.A.; Rand, R.P.; Rau, D.C. Osmotic stress, crowding, preferential hydration, and binding: A comparison of perspectives. Proc. Natl. Acad. Sci. USA 2000, 97, 3987–3992. [Google Scholar] [CrossRef] [Green Version]
- Timasheff, S.N. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc. Natl. Acad. Sci. USA 2002, 99, 9721–9726. [Google Scholar] [CrossRef] [Green Version]
- Licata, V.J.; Allewell, N.M. Measuring hydration changes of proteins in solution: Applications of osmotic stress and structure-based calculations. In Methods in Enzymology; Academic Pres/Elsevier: Cambridge, MA, USA, 1998; Volume 295, pp. 42–62. [Google Scholar]
- Ellis, R.J. Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Minton, A.P. The Influence of Macromolecular Crowding and Macromolecular Confinement on Biochemical Reactions in Physiological Media. J. Biol. Chem. 2001, 276, 10577–10580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.-X.; Rivas, G.; Minton, A.P. Macromolecular Crowding and Confinement: Biochemical, Biophysical, and Potential Physiological Consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, M. Crowding, Diffusion, and Biochemical Reactions. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2014; pp. 383–417. [Google Scholar]
- Dinu, M.V.; Spulber, M.; Renggli, K.; Wu, D.; Monnier, C.A.; Petri-Fink, A.; Bruns, N. Filling polymersomes with polymers by peroxidase-catalyzed atom transfer radical polymerization. Macromol. Rapid Commun. 2015, 36, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Spulber, M.; Fischer, O.; Car, A.; Meier, W. Investigation of Horseradish Peroxidase Kinetics in an “Organelle-Like” Environment. Small 2017, 13, 1603943. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, R. Fractal Reaction Kinetics. Science 1988, 241, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S.; Turner, T.E. Reaction kinetics in intracellular environments with macromolecular crowding: Simulations and rate laws. Prog. Biophys. Mol. Biol. 2004, 85, 235–260. [Google Scholar] [CrossRef] [PubMed]
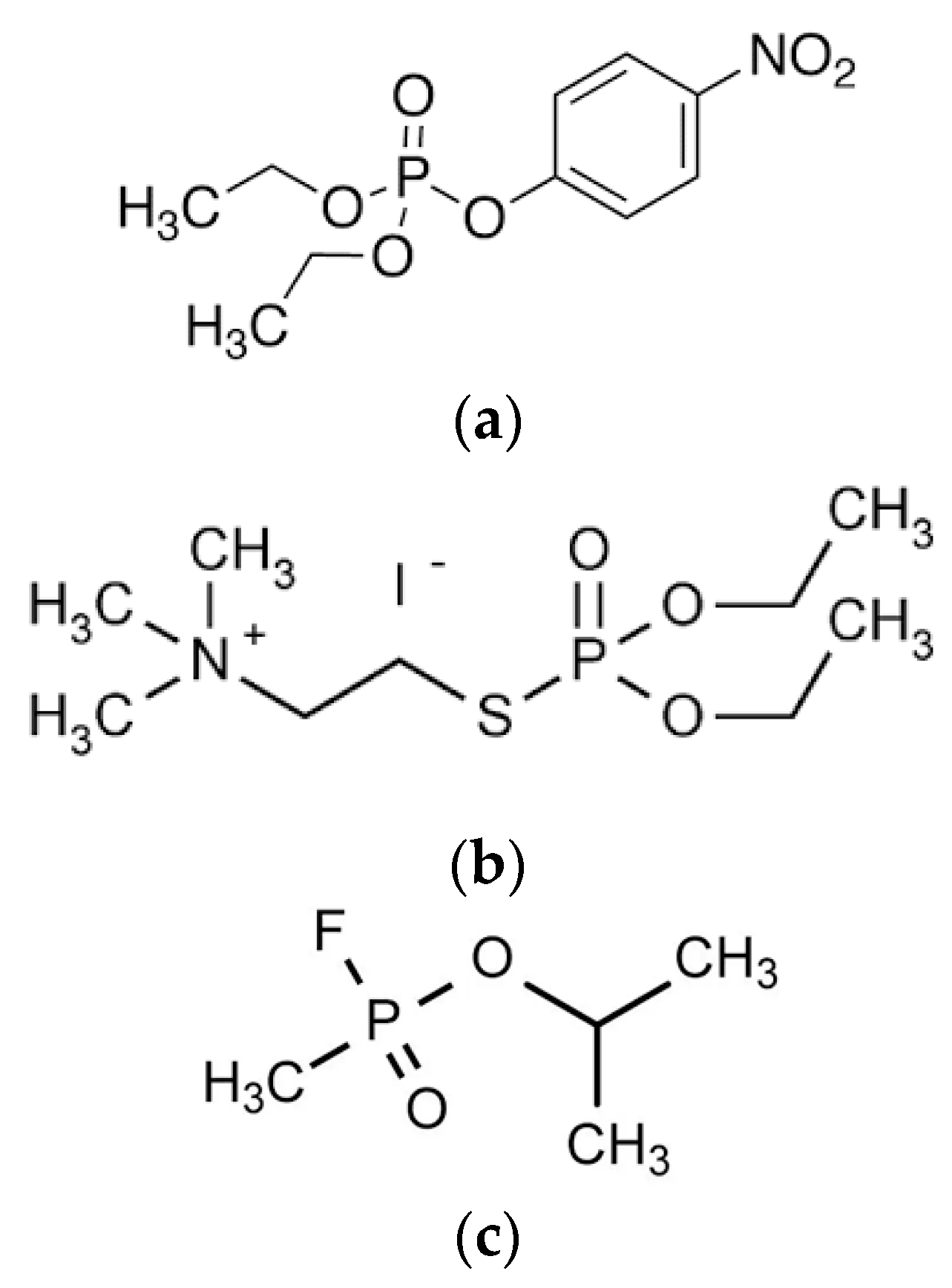
- Zhang, L.; Murata, H.; Amitai, G.; Smith, P.N.; Matyjaszewski, K.; Russell, A.J. Catalytic Detoxification of Organophosphorus Nerve Agents by Butyrylcholinesterase-Polymer-Oxime Bioscavengers. Biomacromolecules 2020, 21, 3867–3877. [Google Scholar] [CrossRef]
- Stenzel, J.; Worek, F.; Eyer, P. Preparation and characterization of dialkylphosphoryl-obidoxime conjugates, potent anticholinesterase derivatives that are quickly hydrolyzed by human paraoxonase (PON1192Q). Biochem. Pharmacol. 2007, 74, 1390–1400. [Google Scholar] [CrossRef]
- Masson, P.; Lushchekina, S.V. Catalytic bioscavengers: The second generation of bioscavenger-based medical countermeasures. In Handbook of Toxicology of Chemical Warfare Agents; Elsevier: Amsterdam, The Netherlands, 2020; Volume 2, pp. 1199–1229. ISBN 9780128190906. [Google Scholar]
- Zueva, I.V.; Lushchekina, S.V.; Daudé, D.; Chabrière, E.; Masson, P. Steady-State Kinetics of Enzyme-Catalyzed Hydrolysis of Echothiophate, a P–S Bonded Organophosphorus as Monitored by Spectrofluorimetry. Molecules 2020, 25, 1371. [Google Scholar] [CrossRef] [Green Version]



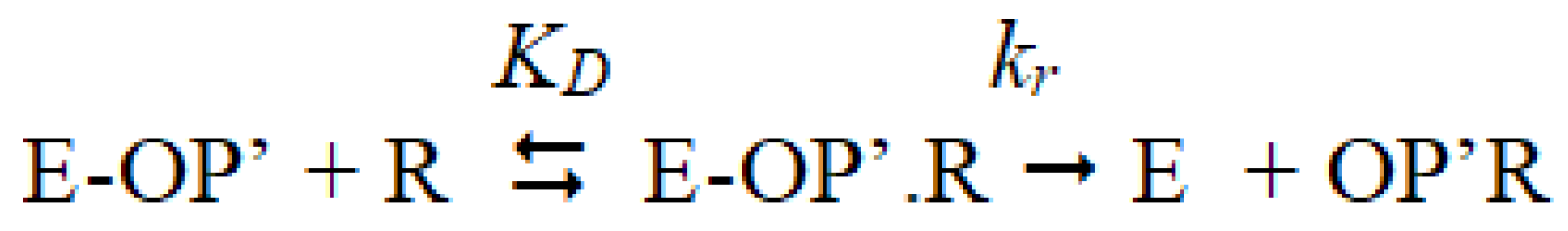




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shajhutdinova, Z.; Pashirova, T.; Masson, P. Kinetic Processes in Enzymatic Nanoreactors for In Vivo Detoxification. Biomedicines 2022, 10, 784. https://doi.org/10.3390/biomedicines10040784
Shajhutdinova Z, Pashirova T, Masson P. Kinetic Processes in Enzymatic Nanoreactors for In Vivo Detoxification. Biomedicines. 2022; 10(4):784. https://doi.org/10.3390/biomedicines10040784
Chicago/Turabian StyleShajhutdinova, Zukhra, Tatiana Pashirova, and Patrick Masson. 2022. "Kinetic Processes in Enzymatic Nanoreactors for In Vivo Detoxification" Biomedicines 10, no. 4: 784. https://doi.org/10.3390/biomedicines10040784
APA StyleShajhutdinova, Z., Pashirova, T., & Masson, P. (2022). Kinetic Processes in Enzymatic Nanoreactors for In Vivo Detoxification. Biomedicines, 10(4), 784. https://doi.org/10.3390/biomedicines10040784