
Comparative Transcriptome Analysis Reveals That Exendin-4 Improves Steatosis in HepG2 Cells by Modulating Signaling Pathways Related to Lipid Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Oleic Acid
2.2. Induction of Steatosis with Oleic Acid
2.3. Treatment with Ex-4
2.4. RNA Extraction
2.5. Library Preparation and RNA Sequencing
2.6. Bioinformatics Analysis
2.7. Functional and Biological Pathway Analysis
3. Results
3.1. Identification of DEGs
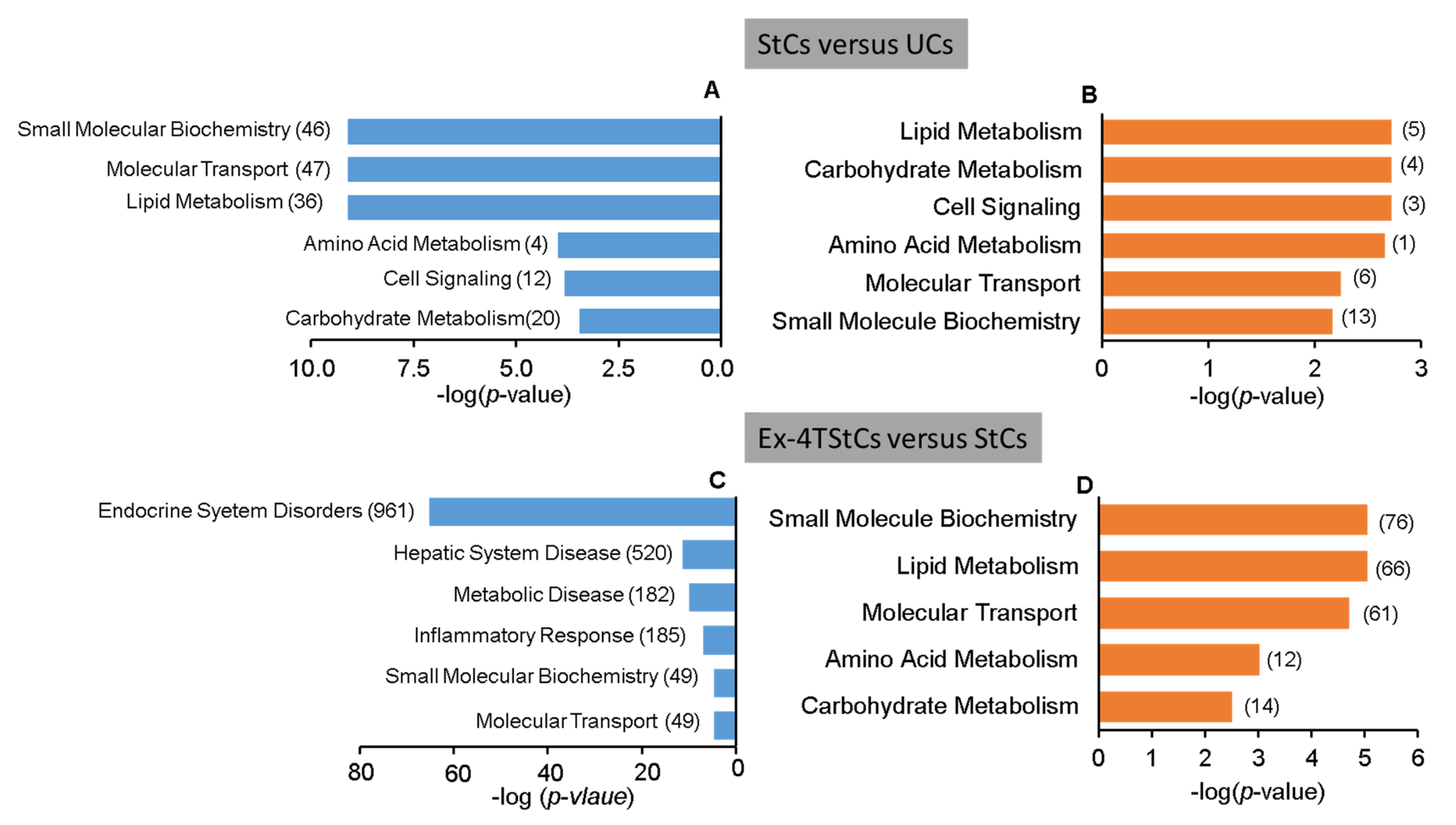
3.2. Steatotic versus Untreated Cells
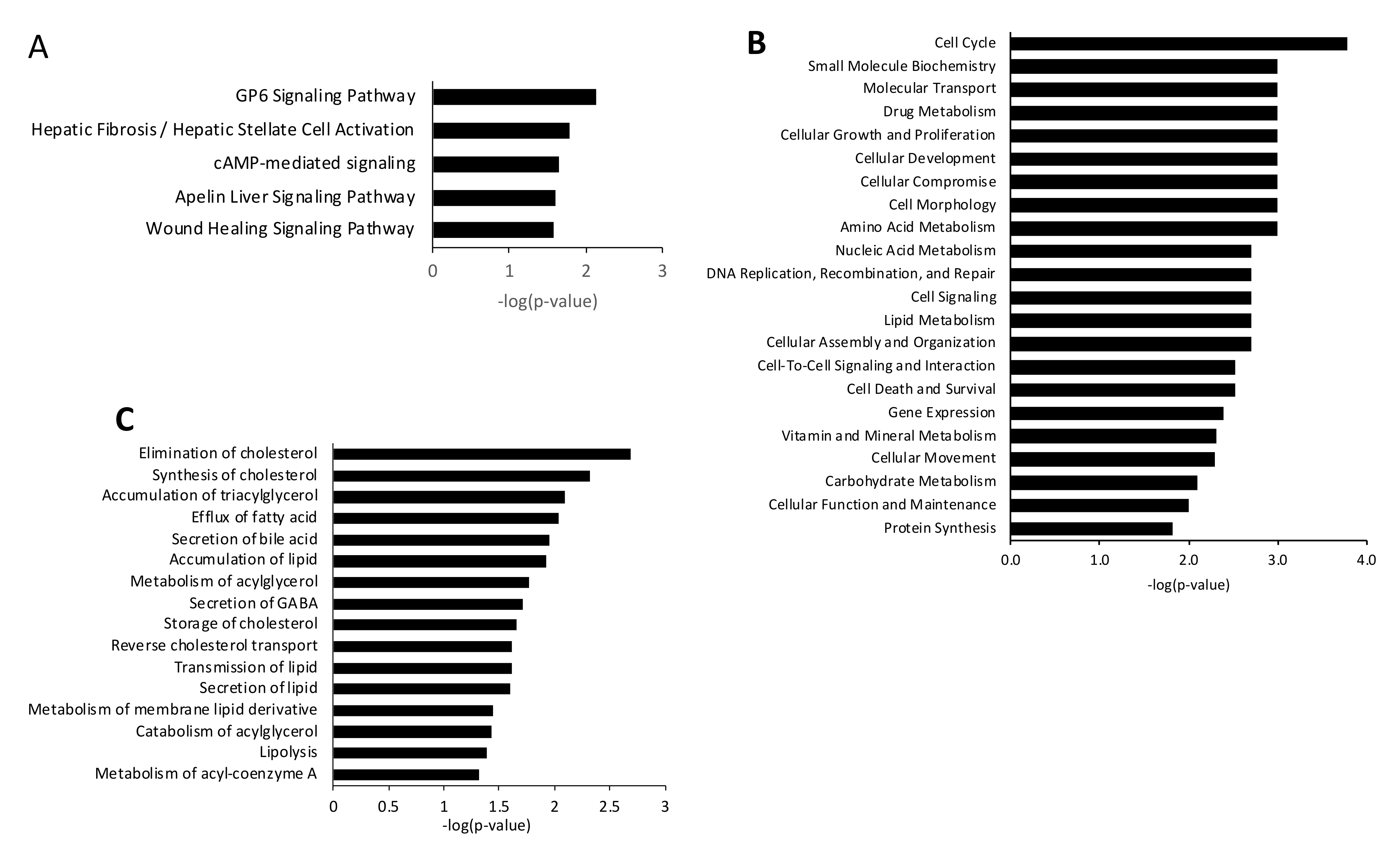
3.3. Steatotic vs. Ex-4-Treated Steatotic Cells
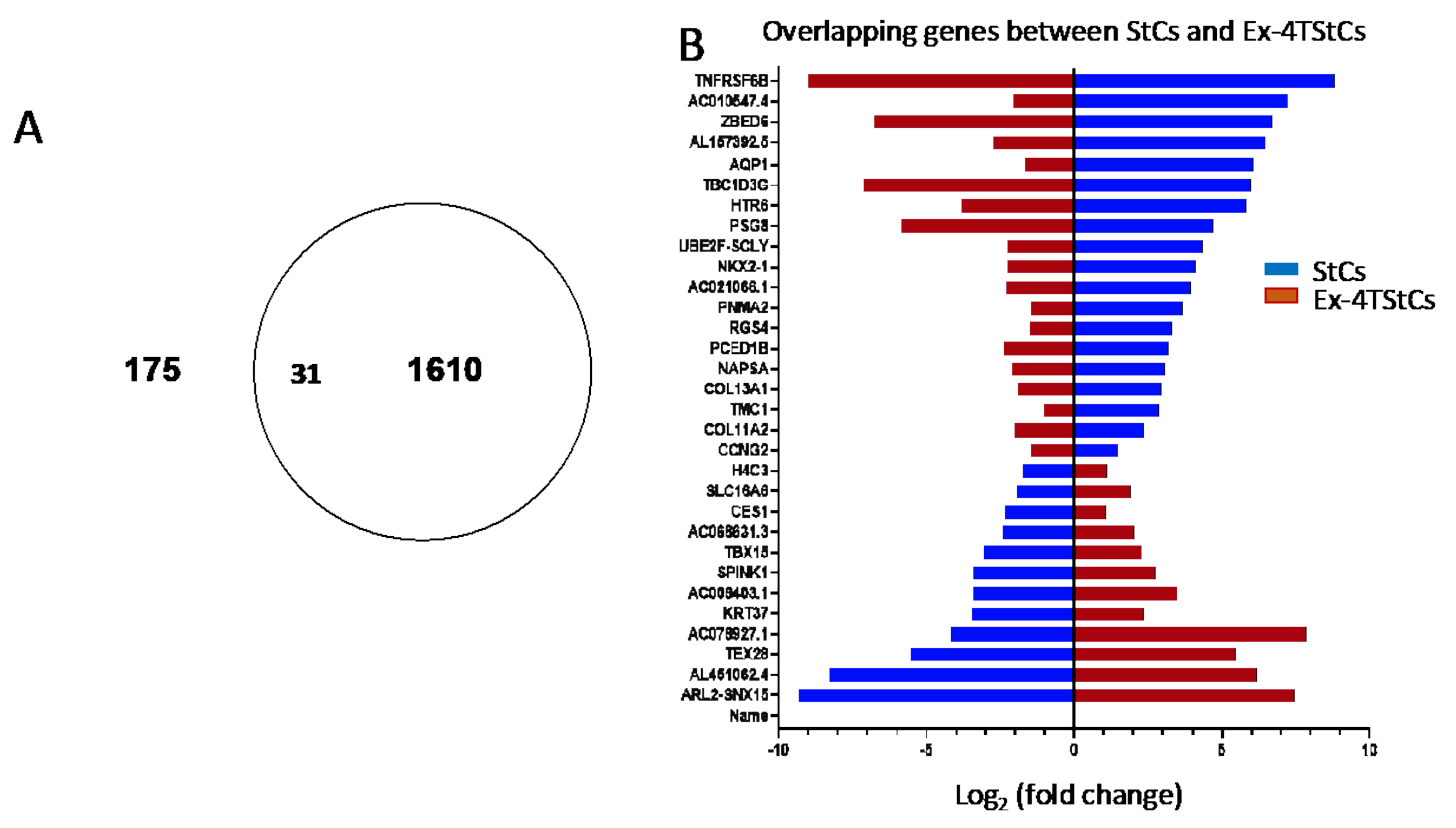
3.4. Overlapping Genes between StCs and Ex-4TStCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Metrakos, P.; Nilsson, T. Non-alcoholic fatty liver disease-a chronic disease of the 21st century. J. Biomed. Res. 2018, 32, 327–335. [Google Scholar] [CrossRef]
- Targher, G.; Tilg, H.; Byrne, C.D. Non-alcoholic fatty liver disease: A multisystem disease requiring a multidisciplinary and holistic approach. Lancet Gastroenterol. Hepatol. 2021, 6, 578–588. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Loomba, R.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; Ratziu, V.; et al. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018, 68, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar]
- Manco, M. Insulin Resistance and Nafld: A Dangerous Liaison beyond the Genetics. Children 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Pang, Q.; Zhang, J.-Y.; Song, S.-D.; Qu, K.; Xu, X.-S.; Liu, S.-S.; Liu, C. Central obesity and nonalcoholic fatty liver disease risk after adjusting for body mass index. World J. Gastroenterol. 2015, 21, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, M.; Yamasandhi, P.G. Nonalcoholic fatty liver disease and Type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2018, 22, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, D.; Georgiopoulos, G.; Katsi, V.; Kourek, C.; Tsioufis, C.; Alexopoulou, A.; Koutli, E.; Tousoulis, D. Non-alcoholic fatty liver disease and hypertension: Coprevalent or correlated? Eur. J. Gastroenterol. Hepatol. 2018, 30, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Q.; Lu, L.-G. Nonalcoholic Fatty Liver Disease: Dyslipidemia, Risk for Cardiovascular Complications, and Treatment Strategy. J. Clin. Transl. Hepatol. 2015, 3, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Kang, E.S.; Boutari, C.; Rhee, E.-J.; Mantzoros, C.S. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism 2020, 111, 154203. [Google Scholar] [CrossRef] [PubMed]
- Hoek, A.M.V.D.; de Jong, J.C.; Worms, N.; van Nieuwkoop, A.; Voskuilen, M.; Menke, A.L.; Lek, S.; Caspers, M.P.; Verschuren, L.; Kleemann, R. Diet and exercise reduce pre-existing NASH and fibrosis and have additional beneficial effects on the vasculature, adipose tissue and skeletal muscle via organ-crosstalk. Metabolism 2021, 124, 154873. [Google Scholar] [CrossRef] [PubMed]
- Katsagoni, C.N.; Papachristou, E.; Sidossis, A.; Sidossis, L. Effects of Dietary and Lifestyle Interventions on Liver, Clinical and Metabolic Parameters in Children and Adolescents with Non-Alcoholic Fatty Liver Disease: A Systematic Review. Nutrients 2020, 12, 2864. [Google Scholar] [CrossRef]
- Meir, A.Y.; Rinott, E.; Tsaban, G.; Zelicha, H.; Kaplan, A.; Rosen, P.; Shelef, I.; Youngster, I.; Shalev, A.; Blüher, M.; et al. Effect of green-Mediterranean diet on intrahepatic fat: The Direct Plus Randomised Controlled Trial. Gut 2021, 70, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.J.; Yu, S.J.; Jung, G.C.; Kwak, M.-S.; Yang, J.I.; Yim, J.Y.; Chung, G.E. Body weight gain rather than body weight variability is associated with increased risk of nonalcoholic fatty liver disease. Sci. Rep. 2021, 11, 14428. [Google Scholar] [CrossRef]
- Friesen, C.S.; Hosey-Cojocari, C.; Chan, S.S.; Csanaky, I.L.; Wagner, J.B.; Sweeney, B.R.; Friesen, A.; Fraser, J.D.; Shakhnovich, V. Efficacy of Weight Reduction on Pediatric Nonalcoholic Fatty Liver Disease: Opportunities to Improve Treatment Outcomes Through Pharmacotherapy. Front. Endocrinol. 2021, 12, 663351. [Google Scholar] [CrossRef]
- Viveiros, K. The Role of Life Style Modifications in Comprehensive Non-Alcoholic Fatty Liver Disease Treatment. Clin. Liver Dis. 2021, 17, 11–14. [Google Scholar] [CrossRef]
- Evert, A.B.; Franz, M.J. Why Weight Loss Maintenance Is Difficult. Diabetes Spectr. 2017, 30, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-Like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef]
- Chavez, C.P.; Cusi, K.; Kadiyala, S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of Nafld. J. Clin. Endocrinol. Metab. 2022, 107, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Lee, M.H.; Yaow, C.Y.L.; Chin, Y.H.; Goh, X.L.; Ng, C.H.; Lim, A.Y.L.; Muthiah, M.D.; Khoo, C.M. Glucagon-Like Peptide-1 Receptor Agonists for Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: A Meta-Analysis. Front. Endocrinol. 2021, 12, 609110. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadkarni, P.; Chepurny, O.G.; Holz, G.G. Regulation of Glucose Homeostasis by GLP-1. Prog. Mol. Biol. Transl. Sci. 2014, 121, 23–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, A.M.; Nguyen, N.Q.; Stevens, J.E.; Fraser, R.J.L.; Holloway, R.H.; Besanko, L.K.; Burgstad, C.; Jones, K.; Chapman, M.J.; Rayner, C.K.; et al. Endogenous Glucagon-Like Peptide-1 Slows Gastric Emptying in Healthy Subjects, Attenuating Postprandial Glycemia. J. Clin. Endocrinol. Metab. 2010, 95, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Vilsbøll, T.; Agersø, H.; Krarup, T.; Holst, J.J. Similar Elimination Rates of Glucagon-Like Peptide-1 in Obese Type 2 Diabetic Patients and Healthy Subjects. J. Clin. Endocrinol. Metab. 2003, 88, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.; Hylleberg, B.; Ng, K.; Damsbo, P. Glucagon-Like Peptide-1 Infusion Must Be Maintained for 24 h/day to Obtain Acceptable Glycemia in Type 2 Diabetic Patients Who Are Poorly Controlled on Sulphonylurea Treatment. Diabetes Care 2001, 24, 1416–1421. [Google Scholar] [CrossRef] [Green Version]
- Hinnen, D. Glucagon-Like Peptide 1 Receptor Agonists for Type 2 Diabetes. Diabetes Spectr. 2017, 30, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; De Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep. 2019, 9, 16810. [Google Scholar] [CrossRef] [PubMed]
- Ard, J.; Fitch, A.; Fruh, S.; Herman, L. Weight Loss and Maintenance Related to the Mechanism of Action of Glucagon-Like Peptide 1 Receptor Agonists. Adv. Ther. 2021, 38, 2821–2839. [Google Scholar] [CrossRef] [PubMed]
- Sofogianni, A.; Filippidis, A.; Chrysavgis, L.; Tziomalos, K.; Cholongitas, E. Glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: An update. World J. Hepatol. 2020, 12, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Ahadi, M.; Molooghi, K.; Masoudifar, N.; Namdar, A.B.; Vossoughinia, H.; Farzanehfar, M. A review of non-alcoholic fatty liver disease in non-obese and lean individuals. J. Gastroenterol. Hepatol. 2021, 36, 1497–1507. [Google Scholar] [CrossRef]
- Errafii, K.; Al-Akl, N.S.; Khalifa, O.; Arredouani, A. Comprehensive analysis of LncRNAs expression profiles in an in vitro model of steatosis treated with Exendin-4. J. Transl. Med. 2021, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Alkhatatbeh, M.J.; Lincz, L.F.; Thorne, R.F. Low simvastatin concentrations reduce oleic acid-induced steatosis in HepG2 cells: An in vitro model of non-alcoholic fatty liver disease. Exp. Ther. Med. 2016, 11, 1487–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, O.; Al-Akl, N.S.; Errafii, K.; Arredouani, A. Exendin-4 alleviates steatosis in an in vitro cell model by lowering FABP1 and FOXA1 expression via the Wnt/-catenin signaling pathway. Sci. Rep. 2022, 12, 2226. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Dong, T.; Saheki, T.; Matsumoto, M.; Iwama, H.; Zhang, H.; et al. Role of ATP-binding cassette transporter A1 in suppressing lipid accumulation by glucagon-like peptide-1 agonist in hepatocytes. Mol. Metab. 2020, 34, 16–26. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.H.; Lee, J.; Hong, S.-W.; Rhee, E.-J.; Park, S.E.; Park, C.Y.; Oh, K.W.; Park, S.W.; Lee, W.-Y. Exendin-4 Inhibits Hepatic Lipogenesis by Increasing β-Catenin Signaling. PLoS ONE 2016, 11, e0166913. [Google Scholar] [CrossRef]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; De Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis inob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Yokomori, H.; Ando, W. Spatial expression of glucagon-like peptide 1 receptor and caveolin-1 in hepatocytes with macrovesicular steatosis in non-alcoholic steatohepatitis. BMJ Open Gastroenterol. 2020, 7, e000370. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Han, J.; Wang, Q.; Mao, Y.; Wei, M.; Jia, W.; Wei, L. A Novel Interacting Protein SERP1 Regulates the N-Linked Glycosylation and Function of GLP-1 Receptor in the Liver. J. Cell. Biochem. 2017, 118, 3616–3626. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Q.; Sun, Y.; Zhao, X.; Kong, Y.; Ou, X.; Jia, J.; Wu, S.; You, H. The Prevalence of Lean/Nonobese Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Clin. Gastroenterol. 2020, 54, 378–387. [Google Scholar] [CrossRef]
- Albhaisi, S.; Chowdhury, A.; Sanyal, A.J. Non-alcoholic fatty liver disease in lean individuals. JHEP Rep. 2019, 1, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mato, J.M.; Alonso, C.; Noureddin, M.; Lu, S.C. Biomarkers and subtypes of deranged lipid metabolism in non-alcoholic fatty liver disease. World J. Gastroenterol. 2019, 25, 3009–3020. [Google Scholar] [CrossRef]
- Jiao, Y.; Lu, Y.; Li, X.-Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Panzitt, K.; Wagner, M. FXR in liver physiology: Multiple faces to regulate liver metabolism. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166133. [Google Scholar] [CrossRef]
- Jadhav, K.; Xu, Y.; Xu, Y.; Li, Y.; Xu, J.; Zhu, Y.; Adorini, L.; Lee, Y.K.; Kasumov, T.; Yin, L.; et al. Reversal of metabolic disorders by pharmacological activation of bile acid receptors TGR5 and FXR. Mol. Metab. 2018, 9, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Pettinelli, P.; Videla, L.A. Up-Regulation of Ppar-Gamma Mrna Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to Srebp-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Hajri, T.; Zaiou, M.; Fungwe, T.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARγ expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef] [PubMed]
- Decara, J.; Arrabal, S.; Beiroa, D.; Rivera, P.; Vargas, A.; Serrano, A.; Pavón, F.J.; Ballesteros, J.; Dieguez, C.; Nogueiras, R. Antiobesity Efficacy of Glp-1 Receptor Agonist Liraglutide Is Associated with Peripheral Tissue-Specific Modulation of Lipid Metabolic Regulators. Biofactors 2016, 42, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Huang, Y.; Yan, L.; Gao, M.; Liu, D. Synthetic FXR Agonist GW4064 Prevents Diet-Induced Hepatic Steatosis and Insulin Resistance. Pharm. Res. 2013, 30, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.-H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.-L.M.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased Hepatic CD36 Expression Contributes to Dyslipidemia Associated With Diet-Induced Obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Distrutti, E.; Santucci, L.; Cipriani, S.; Renga, B.; Schiaroli, E.; Ricci, P.; Donini, A.; Fiorucci, S. Bile acid activated receptors are targets for regulation of integrity of gastrointestinal mucosa. J. Gastroenterol. 2015, 50, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.S.; Reddy, J.K. Peroxisomal Beta-Oxidation and Steatohepatitis. Semin Liver Dis. 2001, 21, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Jia, Y.; Yang, G.; Zhang, X.; Boddu, P.C.; Petersen, B.; Narsingam, S.; Zhu, Y.-J.; Thimmapaya, B.; Kanwar, Y.S.; et al. PPARα-Deficient ob/ob Obese Mice Become More Obese and Manifest Severe Hepatic Steatosis Due to Decreased Fatty Acid Oxidation. Am. J. Pathol. 2015, 185, 1396–1408. [Google Scholar] [CrossRef] [Green Version]
- Zelcer, N.; Tontonoz, P. Liver X Receptors as Integrators of Metabolic and Inflammatory Signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Lima-Cabello, E.; García-Mediavilla, M.V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lozano-Rodríguez, T.; Fernández-Bermejo, M.; Olcoz, J.L.; González-Gallego, J.; García-Monzón, C.; Sánchez-Campos, S. Enhanced Expression of Pro-Inflammatory Mediators and Liver X-Receptor-Regulated Lipogenic Genes in Non-Alcoholic Fatty Liver Disease and Hepatitis C. Clin. Sci. 2011, 120, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. Srebps: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Wouters, K.; Van Bilsen, M.; van Gorp, P.; Bieghs, V.; Lütjohann, D.; Kerksiek, A.; Staels, B.; Hofker, M.H.; Shiri-Sverdlov, R. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett. 2010, 584, 1001–1005. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Han, X.; Bian, Z.; Peng, Y.; You, Z.; Wang, Q.; Chen, X.; Qiu, D.; Ma, X. Activation of Liver X Receptors Attenuates Endotoxin-Induced Liver Injury in Mice with Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2011, 57, 390–398. [Google Scholar] [CrossRef]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.S.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 2015, 4, e08009. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Errafii, K.; Khalifa, O.; Al-Akl, N.S.; Arredouani, A. Comparative Transcriptome Analysis Reveals That Exendin-4 Improves Steatosis in HepG2 Cells by Modulating Signaling Pathways Related to Lipid Metabolism. Biomedicines 2022, 10, 1020. https://doi.org/10.3390/biomedicines10051020
Errafii K, Khalifa O, Al-Akl NS, Arredouani A. Comparative Transcriptome Analysis Reveals That Exendin-4 Improves Steatosis in HepG2 Cells by Modulating Signaling Pathways Related to Lipid Metabolism. Biomedicines. 2022; 10(5):1020. https://doi.org/10.3390/biomedicines10051020
Chicago/Turabian StyleErrafii, Khaoula, Olfa Khalifa, Neyla S. Al-Akl, and Abdelilah Arredouani. 2022. "Comparative Transcriptome Analysis Reveals That Exendin-4 Improves Steatosis in HepG2 Cells by Modulating Signaling Pathways Related to Lipid Metabolism" Biomedicines 10, no. 5: 1020. https://doi.org/10.3390/biomedicines10051020