
The Tarantula Toxin ω-Avsp1a Specifically Inhibits Human CaV3.1 and CaV3.3 via the Extracellular S3-S4 Loop of the Domain 1 Voltage-Sensor

 ,
,  , ,
, , 
Abstract
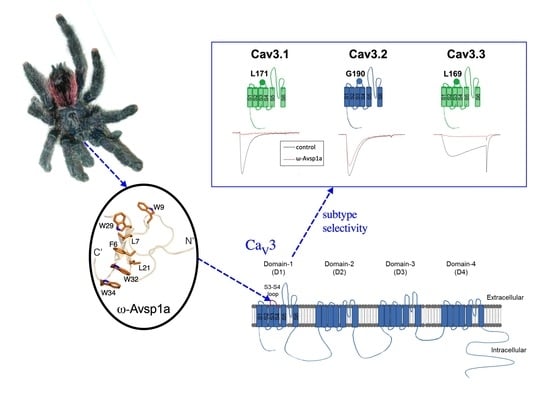
:
1. Introduction
2. Materials and Methods
2.1. Venoms
2.2. Reversed-Phase High Pressure Liquid Chromatography
2.3. Mass Spectrometry
2.4. N-Terminal Edman Sequencing
2.5. C-Terminal Sequencing Using Carboxypeptidase Y
2.6. Chemical Synthesis of ω-Avsp1a
2.7. Cell Culture and Transient Expression
2.8. Electrophysiology
2.9. Construction and Mutagenesis of Plasmid cDNA
2.10. Determination of Three-Dimensional (3D) Structure of ω-Avsp1a by NMR
3. Results
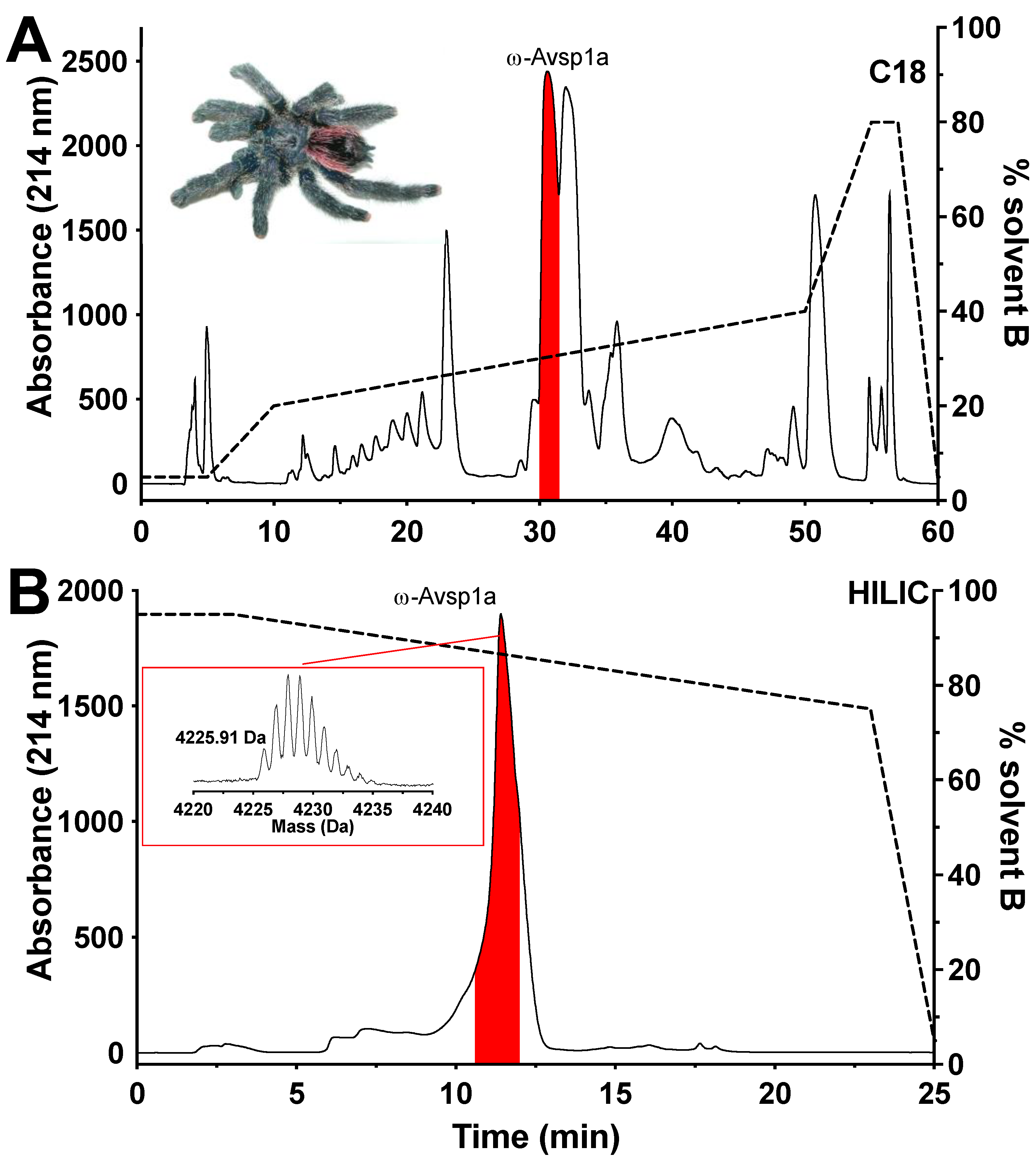
3.1. Toxin Isolation and Purification
3.2. Chemical Synthesis of ω-Avsp1a
3.3. Sub-Type Specific Inhibition of T-Type Calcium Channels by ω-Avsp1a
3.4. Identification of T-Type Calcium Channel α1-Subunit Domains Involved in ω-Avsp1a Activity
3.5. Identification of ω-Avsp1a Binding Site on T-Type Calcium Channels
3.6. Quantification of the Potency of Synthetic ω-Avsp1a against CaV3.1 and CaV3.3
3.7. The 3D Structure of ω-Avsp1a
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Missiaen, L.; Callewaert, G.; Parys, J.B.; Wuytack, F.; Raeymaekers, L.; Droogmans, G.; Nilius, B.; Eggermont, J.; De Smedt, H. Intracellular calcium: Physiology and physiopathology. Verh.-K. Acad. Geneeskd. Belg. 2000, 62, 471–499. [Google Scholar] [PubMed]
- Missiaen, L.; Robberecht, W.; van den Bosch, L.; Callewaert, G.; Parys, J.B.; Wuytack, F.; Raeymaekers, L.; Nilius, B.; Eggermont, J.; De Smedt, H. Abnormal intracellular Ca2+homeostasis and disease. Cell Calcium 2000, 28, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Perez-Reyes, E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Song, I.; Keum, S.; Lee, T.; Jeong, M.J.; Kim, S.S.; McEnery, M.W.; Shin, H.S. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α(1G) T-type Ca2+ channels. Neuron 2001, 31, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Lamping, K.G.; Nuno, D.W.; Barresi, R.; Prouty, S.J.; Lavoie, J.L.; Cribbs, L.L.; England, S.K.; Sigmund, C.D.; Weiss, R.M.; et al. Abnormal coronary function in mice deficient in α1H T-type Ca2+ channels. Science 2003, 302, 1416–1418. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, B.H.; Chen, Y.H.; Huang, C.H.; Lin, S.S.; Lee, K.R.; Chen, C.C. The CaV3.1 T-type calcium channel is required for neointimal formation in response to vascular injury in mice. Cardiovasc. Res. 2012, 96, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.E.; Traboulsie, A.; Leoni, A.L.; Couette, B.; Marger, L.; Le Quang, K.; Kupfer, E.; Cohen-Solal, A.; Vilar, J.; Shin, H.S.; et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-type calcium channels. Circ. Res. 2006, 98, 1422–1430. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.P.; Mochizuki, T.; Xie, J.; Fischler, W.; Manger, J.P.; Talley, E.M.; Scammell, T.E.; Tonegawa, S. Thalamic CaV3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc. Natl. Acad. Sci. USA 2005, 102, 1743–1748. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, D.; Shin, H.S. Lack of delta waves and sleep disturbances during non-rapid eye movement sleep in mice lacking α1G-subunit of T-type calcium channels. Proc. Natl. Acad. Sci. USA 2004, 101, 18195–18199. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.S.; Huang, C.H.; Chieng, H.; Chang, Y.T.; Chang, D.; Chen, J.J.; Chen, Y.C.; Chen, Y.H.; Shin, H.S.; Campbell, K.P.; et al. The CaV3.2 T-type Ca2+ channel is required for pressure overload-induced cardiac hypertrophy in mice. Circ. Res. 2009, 104, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Bourinet, E.; Alloui, A.; Monteil, A.; Barrere, C.; Couette, B.; Poirot, O.; Pages, A.; McRory, J.; Snutch, T.P.; Eschalier, A.; et al. Silencing of the CaV3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005, 24, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Caballero, A.; Gadotti, V.M.; Stemkowski, P.; Weiss, N.; Souza, I.A.; Hodgkinson, V.; Bladen, C.; Chen, L.; Hamid, J.; Pizzoccaro, A.; et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing CaV3.2 channel activity. Neuron 2014, 83, 1144–1158. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.K.; Liu, I.Y.; Chang, Y.T.; Chen, Y.C.; Chen, C.C.; Yen, C.T.; Shin, H.S.; Chen, C.C. CaV3.2 T-type Ca2+ channel-dependent activation of ERK in paraventricular thalamus modulates acid-induced chronic muscle pain. J. Neurosci. 2010, 30, 10360–10368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, P.B. Functional importance of T-type voltage-gated calcium channels in the cardiovascular and renal system: News from the world of knockout mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R227–R237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Bodi, I.; Correll, R.N.; Chen, X.W.; Lorenz, J.; Houser, S.R.; Robbins, J.; Schwartz, A.; Molkentin, J.D. α1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J. Clin. Investig. 2009, 119, 3787–3796. [Google Scholar] [CrossRef] [PubMed]
- Bourinet, E.; Zamponi, G.W. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology 2017, 127, 109–115. [Google Scholar] [CrossRef]
- Norton, R.S.; McDonough, S.I. Peptides targeting voltage-gated calcium channels. Curr. Pharm. Des. 2008, 14, 2480–2491. [Google Scholar] [CrossRef]
- Cardoso, F.C. Multi-targeting sodium and calcium channels using venom peptides for the treatment of complex ion channels-related diseases. Biochem. Pharmacol. 2020, 181, 114107. [Google Scholar] [CrossRef]
- King, G.F. Modulation of insect CaV channels by peptidic spider toxins. Toxicon 2007, 49, 513–530. [Google Scholar] [CrossRef]
- Pringos, E.; Vignes, M.; Martinez, J.; Rolland, V. Peptide neurotoxins that affect voltage-gated calcium channels: A close-up on ω-agatoxins. Toxins 2011, 3, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Herzig, V.; Hodgson, W.C. Intersexual variations in the pharmacological properties of Coremiocnemis tropix (Araneae, Theraphosidae) spider venom. Toxicon 2009, 53, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.W.; Park, J.Y.; Jeong, S.W.; Kim, J.A.; Moon, H.J.; Perez-Reyes, E.; Lee, J.H. A molecular determinant of nickel inhibition in CaV3.2 T-type calcium channels. J. Biol. Chem. 2006, 281, 4823–4830. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.T.; Woo, J.; Kang, H.W.; Vitko, I.; Barrett, P.Q.; Perez-Reyes, E.; Lee, J.H.; Shin, H.S.; Todorovic, S.M. Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J. Neurosci. 2007, 27, 8250–8260. [Google Scholar] [CrossRef] [Green Version]
- King, G.F.; Hardy, M.C. Spider-venom peptides: Structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef]
- Pineda, S.S.; Chin, Y.K.; Undheim, E.A.B.; Senff, S.; Mobli, M.; Dauly, C.; Escoubas, P.; Nicholson, G.M.; Kaas, Q.; Guo, S.; et al. Structural venomics reveals evolution of a complex venom by duplication and diversification of an ancient peptide-encoding gene. Proc. Natl. Acad. Sci. USA 2020, 117, 11399–11408. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Herzig, V.; King, G.F. The cystine knot is responsible for the exceptional stability of the insecticidal spider toxin ω-Hexatoxin-Hv1a. Toxins 2015, 7, 4366–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, C.Y.; Chin, Y.K.Y.; Ma, L.; Undheim, E.A.B.; Herzig, V.; King, G.F. A selective NaV1.1 activator with potential for treatment of Dravet syndrome epilepsy. Biochem. Pharmacol. 2020, 181, 113991. [Google Scholar] [CrossRef]
- Ma, L.; Chin, Y.K.Y.; Dekan, Z.; Herzig, V.; Chow, C.Y.; Heighway, J.; Lam, S.W.; Guillemin, G.J.; Alewood, P.F.; King, G.F. Novel venom-derived inhibitors of the human EAG channel, a putative antiepileptic drug target. Biochem. Pharmacol. 2018, 158, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Redd, M.A.; Scheuer, S.E.; Saez, N.J.; Yoshikawa, Y.; Chiu, H.S.; Gao, L.; Hicks, M.; Villanueva, J.E.; Joshi, Y.; Chow, C.Y.; et al. Therapeutic inhibition of acid-sensing ion channel 1a recovers heart function after ischemia-reperfusion injury. Circulation 2021, 144, 947–960. [Google Scholar] [CrossRef]
- Deplazes, E.; Henriques, S.T.; Smith, J.J.; King, G.F.; Craik, D.J.; Mark, A.E.; Schroeder, C.I. Membrane-binding properties of gating modifier and pore-blocking toxins: Membrane interaction is not a prerequisite for modification of channel gating. Biochim. Biophys. Acta 2016, 1858, 872–882. [Google Scholar] [CrossRef]
- Henriques, S.T.; Deplazes, E.; Lawrence, N.; Cheneval, O.; Chaousis, S.; Inserra, M.; Thongyoo, P.; King, G.F.; Mark, A.E.; Vetter, I.; et al. Interaction of tarantula venom peptide ProTx-II with lipid membranes Is a prerequisite for its inhibition of human voltage-gated sodium channel NaV1.7. J. Biol. Chem. 2016, 291, 17049–17065. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Miller, W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 1991, 12, 337–357. [Google Scholar] [CrossRef] [Green Version]
- Powell, K.L.; Cain, S.M.; Snutch, T.P.; O’Brien, T.J. Low threshold T-type calcium channels as targets for novel epilepsy treatments. Br. J. Clin. Pharmacol. 2014, 77, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Snutch, T.P.; Zamponi, G.W. Recent advances in the development of T-type calcium channel blockers for pain intervention. Br. J. Pharmacol. 2018, 175, 2375–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Huang, G.; Wu, Q.; Wu, K.; Li, R.; Lei, J.; Pan, X.; Yan, N. Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature 2019, 576, 492–497. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID | 7LVN |
|---|---|
| Experimental restraints | |
| Inter-proton distance restraints | |
| Total | 565 |
| Intra-residue (i = j) | 137 |
| Sequential (|i − j| = 1) | 178 |
| Medium range (1 < |i − j| < 5) | 88 |
| Long range (|i − j| ≥ 5) | 162 |
| Disulfide bond restraints | 9 |
| Dihedral-angle restraints | 52 |
| φ dihedral angle restraints | 23 |
| ψ dihedral angle restraints | 21 |
| χ1 angle restraints | 8 |
| Total number of restraints per residue | 17.4 |
| Violations of experimental restraints | 0 |
| RMSD from mean coordinate structure (Å) b | |
| All backbone atoms | 0.43 ± 0.10 |
| All heavy atoms | 0.74 ± 0.12 |
| Backbone atoms (Residue 3–34) | 0.16 ± 0.06 |
| Heavy atoms (Residue 3–34) | 0.63 ± 0.11 |
| Stereochemical quality c | |
| Ramachandran plot statistics | |
| Residues in most favoured Ramachandran region (%) | 76.0 ± 2.6 |
| Disallowed regions [%] | 2.4 ± 1.2 |
| Unfavourable sidechain rotamers [%] | 7.8 ± 1.9 |
| Clashscore, all atoms d | 0.0 ± 0.0 |
| Overall MolProbity score | 1.95 ± 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herzig, V.; Chen, Y.-C.; Chin, Y.K.-Y.; Dekan, Z.; Chang, Y.-W.; Yu, H.-M.; Alewood, P.F.; Chen, C.-C.; King, G.F. The Tarantula Toxin ω-Avsp1a Specifically Inhibits Human CaV3.1 and CaV3.3 via the Extracellular S3-S4 Loop of the Domain 1 Voltage-Sensor. Biomedicines 2022, 10, 1066. https://doi.org/10.3390/biomedicines10051066
Herzig V, Chen Y-C, Chin YK-Y, Dekan Z, Chang Y-W, Yu H-M, Alewood PF, Chen C-C, King GF. The Tarantula Toxin ω-Avsp1a Specifically Inhibits Human CaV3.1 and CaV3.3 via the Extracellular S3-S4 Loop of the Domain 1 Voltage-Sensor. Biomedicines. 2022; 10(5):1066. https://doi.org/10.3390/biomedicines10051066
Chicago/Turabian StyleHerzig, Volker, Yong-Cyuan Chen, Yanni K.-Y. Chin, Zoltan Dekan, Yu-Wang Chang, Hui-Ming Yu, Paul F. Alewood, Chien-Chang Chen, and Glenn F. King. 2022. "The Tarantula Toxin ω-Avsp1a Specifically Inhibits Human CaV3.1 and CaV3.3 via the Extracellular S3-S4 Loop of the Domain 1 Voltage-Sensor" Biomedicines 10, no. 5: 1066. https://doi.org/10.3390/biomedicines10051066
APA StyleHerzig, V., Chen, Y.-C., Chin, Y. K.-Y., Dekan, Z., Chang, Y.-W., Yu, H.-M., Alewood, P. F., Chen, C.-C., & King, G. F. (2022). The Tarantula Toxin ω-Avsp1a Specifically Inhibits Human CaV3.1 and CaV3.3 via the Extracellular S3-S4 Loop of the Domain 1 Voltage-Sensor. Biomedicines, 10(5), 1066. https://doi.org/10.3390/biomedicines10051066