
Adeno-Associated Viruses for Modeling Neurological Diseases in Animals: Achievements and Prospects


Abstract
:1. Introduction
Recombinant Adeno-Associated Viruses (rAAVs)
1.1.1. AAV Expression Cassette
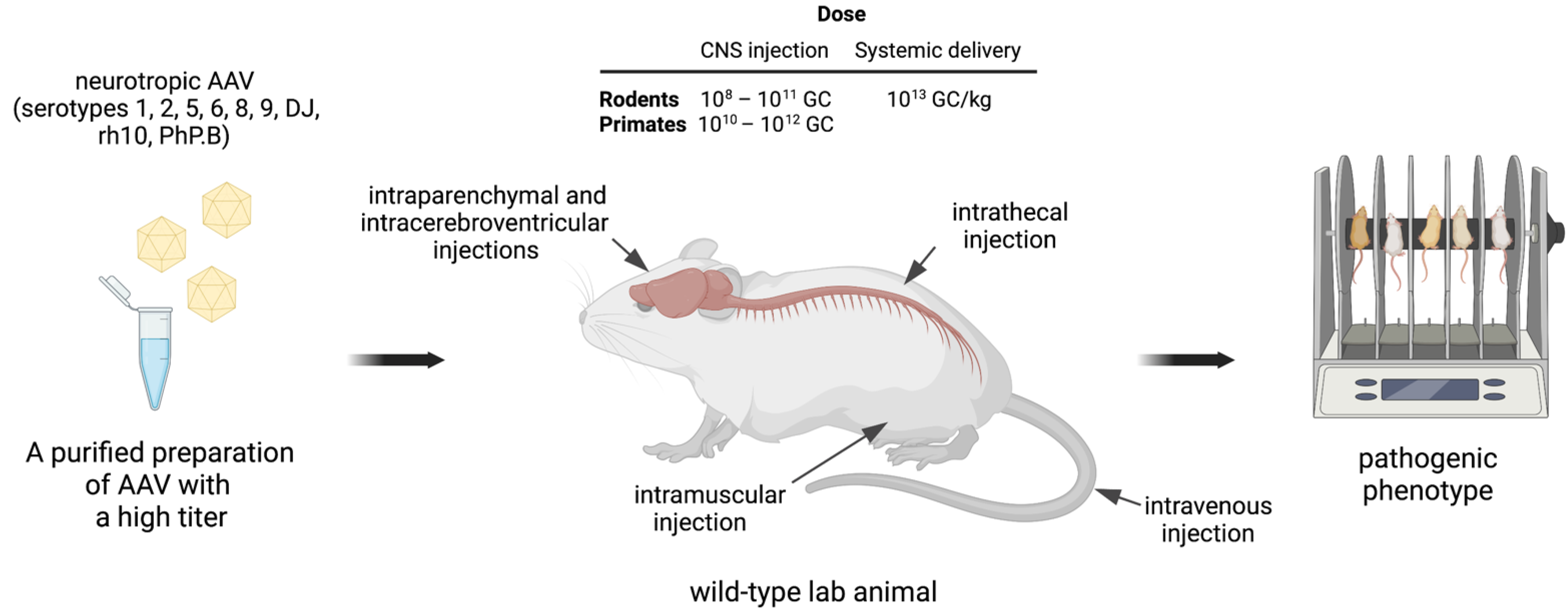
1.1.2. Targeting Neuronal Tissues

{kind=link}
{kind=link}
{kind=link}
| Disease | The Most Affected Area | Animals | Age of Animals * | rAAV Serotype | Dose ** | Injection Route *** | Reference |
|---|---|---|---|---|---|---|---|
| Alzheimer’s disease | Hippocampus, the associative cortex and subcortical structures | Mouse | adult | AAV1 | 1 × 109 TU; 2 μL | IP, hippocampus | [50] |
| Mouse | adult | AAVrh10 | 5 × 108–109 GC/side; 2 μL | IP, hippocampus | [49] | ||
| Rat | adult | AAV1 | 3 × 1010 GC/side; 3 μL | IP, hippocampus | [24] | ||
| Rat | adult | AAV9 | 3 × 109 GC; 3 μL | IP, substantia nigra | [44] | ||
| NHP | adult | AAV1 | 1.176 × 1013 GC/μL sample used, injection volume not specified | IP, entorhinal cortex | [54] | ||
| Parkinson’s disease | Substantia nigra | Rat | adult | AAV6 | 2 × 1010 GC; 2 μL | IP, vagus nerve | [55] |
| Rat | adult | AAV6 | 2.5 × 1010 GC/side; 4 μL | IP, substantia nigra pars compacta | [43] | ||
| Amyotrophic lateral sclerosis, frontotemporal dementia | Motor neurons, Frontal and temporal lobes | Mouse | neonatal | AAV9 | 4.5 × 1010 GC; 5 μL | IT, cisterna magna | [19] |
| Mouse | neonatal | AAV9 | 3 × 1010 GC/side; 2 μL | ICV | [20] | ||
| Amyotrophic lateral sclerosis | Motor neurons | Rat | neonatal adult adult | AAV9 AAV-PHP.B AAV9, AAV-PHP.B AAV9 | 6.7 × 1014 GC/kg 2.7 × 1014 GC/kg 3 × 1013 GC/kg ~1.4 × 1010 GC | IV ICV | [51] |
| Rat | adult | AAV9 | 1–6 × 1013 GC/kg | IV | [56] | ||
| Rat | adult | AAV1 | 4.5 × 109 GC; 1.5 μL | IP, C6 of the spinal cord | [23] | ||
| NHP | adult | AAV1 | 1.5 × 1010 GC or 1.5 × 1011 GC; 5 μL | IP, C5–6 of the spinal cord | [23] | ||
| Huntington’s disease | Striatum | Mouse | juvenile | AAV-DJ | 1.24–2.46 × 109 GC/side; 2 μL | IP, striatum mid-coronal level | [47] |
| Mouse | adult | AAV1/8 mosaic vector | 3.15 or 5.4 × 1010 GC/side; 3 μL | IP, striatum | [45] | ||
| Mouse | adult | AAV5 | 3.45- 7 × 1010 GC/side; 0.5 μL | IP, hypothalamus | [57] | ||
| Mouse | adult | AAV6 | 2.4 × 109 VP/side; 2 μL | IP, layer V of the cortex | [16] | ||
| Rat | neonatal | AAV5 | 1.4 × 1011 GC/side; 1 μL | IP, dorsal striatum | [46] | ||
| Rat | juvenile | AAV9 | 1.52 × 1011 GC/side; 4 μL | IP, striatum | [58] | ||
| Rat | adult | AAV2 | 4 × 109 or 4 × 1010 GC; 2 μL | IP, striatum | [48] | ||
| Rat | adult | chimeric AAV1/AAV2 | 3 × 109 GC/side; 3 μL | IP, striatum | [59] | ||
| NHP | adult | AAV6 | 5.3 × 1012 VP; 200 μL/4 sites | IP, striatum | [16] | ||
| KCNMA1-related cerebellar ataxia **** | Cerebellum (Purkinje cells) | Mouse | neonatal | AAV9 | 2 × 1012 GC/side; 2 μL | ICV | [22] |
| Spinocerebellar ataxia type 6 | Cerebellum (Purkinje cells), spinal cord | Mouse | neonatal | AAV9 | 1 × 1010 GC; 2–4 μL | ICV | [17] |
| Spinocerebellar ataxia type 1 and 3 | Mouse | juvenile | AAV-PHP.B and AAV-PHP.eB | ~6.5 × 1013 GC/kg | IV | [37] | |
| Spinal muscular atrophy | Anterior horns of the spinal cord | Pig | neonatal | AAV9 | 6.5 × 1012 GC/kg | IT, cisterna magna | [31] |
| Goucher disease type 3 | unknown | Mouse | neonatal | AAV1 | 2 × 1010 VP/side; 2 μL | ICV | [32] |
| GNAO1-encephalopathy | unknown | Mouse | adult | AAV9 | 3 × 109 GC/side; 300 nL | IP, dorsal striatum | [21] |
| Focal neocortical epilepsies | Neocortex | Mouse | adult | AAV8 | 5 × 108 GC; 0.5 μL | IP, cortex | [26] |
| Refractory epilepsy | Hippocampus and many other regions | Mouse | adult | AAV8 | 2 × 109 GC; 2 μL | IP, CA3 region of the hippocampus | [25] |
| Major depressive disorder | Amygdala, hippocampus and many other regions | Mouse | juvenile | AAV9 | 5 × 108 GC/side; 0.5 μL | IP, ventral hippocampal dentate gyrus | [60] |
| Mouse | adult | AAV9 | 2.16 × 109 GC; 7.9 × 108 GC/side; 0.2 μL | IP, basolateral amygdala | [28] | ||
| Mouse | adult | AAV8 | 1 × 1011 GC/side; 25 μL | IM, quadriceps | [27] | ||
| Mouse | adult | AAV2 | 5 × 109 GC/side; 0.5 μL | IP, basolateral amygdala | [61] | ||
| Rat | adult | not specified | not specified | IP, dorsal hippocampal dentate gyrus | [62] | ||
| Rat | adult | AAV9 | 1–1.5 × 109 TU; 1–1.5 μL | IP, CA1 region of hippocampus | [63] | ||
| Rat | not specified | not specified | 3 µL of virus, titer not specified | IP, prefrontal cortex | [64] |
2. Examples of Modeling Neurological Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
2.3. Amyotrophic Lateral Sclerosis
2.4. Huntington’s Disease
2.5. Spinocerebellar Ataxias
2.6. Spinal Muscular Atrophy (SMA)
2.7. Lysosomal Storage Diseases
2.8. Epilepsy
2.9. Major Depressive Disorder
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Prim. 2018, 4, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-Ethnic Carrier Screening and Prenatal Diagnosis for Spinal Muscular Atrophy: Clinical Laboratory Analysis of >72 400 Specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef]
- Gurumurthy, C.B.; Kent Lloyd, K.C. Generating Mouse Models for Biomedical Research: Technological Advances. Dis. Model. Mech. 2019, 12, dmm029462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nectow, A.R.; Nestler, E.J. Viral Tools for Neuroscience. Nat. Rev. Neurosci. 2020, 21, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-Associated Virus Vector as a Platform for Gene Therapy Delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Deverman, B.E.; Matho, K.S.; Cetin, A.; Woodard, K.; Cepko, C.; Guerin, K.I.; Rego, M.A.; Ersing, I.; Bachle, S.M.; et al. Adeno-Associated Virus Technologies and Methods for Targeted Neuronal Manipulation. Front. Neuroanat. 2019, 13, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601. [Google Scholar] [CrossRef]
- Hocquemiller, M.; Giersch, L.; Audrain, M.; Parker, S.; Cartier, N. Adeno-Associated Virus-Based Gene Therapy for CNS Diseases. Hum. Gene Ther. 2016, 27, 478–496. [Google Scholar] [CrossRef] [Green Version]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [Green Version]
- Biselli, T.; Lange, S.S.; Sablottny, L.; Steffen, J.; Walther, A. Optogenetic and Chemogenetic Insights into the Neurocircuitry of Depression-like Behaviour: A Systematic Review. Eur. J. Neurosci. 2021, 53, 9–38. [Google Scholar] [CrossRef]
- Haggerty, D.L.; Grecco, G.G.; Reeves, K.C.; Atwood, B. Adeno-Associated Viral Vectors in Neuroscience Research. Mol. Ther. Methods Clin. Dev. 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Bedbrook, C.N.; Deverman, B.E.; Gradinaru, V. Viral Strategies for Targeting the Central and Peripheral Nervous Systems. Annu. Rev. Neurosci. 2018, 41, 323–348. [Google Scholar] [CrossRef]
- Davidsson, M.; Negrini, M.; Hauser, S.; Svanbergsson, A.; Lockowandt, M.; Tomasello, G.; Manfredsson, F.P.; Heuer, A. A Comparison of AAV-Vector Production Methods for Gene Therapy and Preclinical Assessment. Sci. Reports 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.K.; Rivera-Soto, R.; Gray, S.J. Viral Expression Cassette Elements to Enhance Transgene Target Specificity and Expression in Gene Therapy. Discov. Med. 2015, 19, 49. [Google Scholar] [PubMed]
- Maxan, A.; Sciacca, G.; Alpaugh, M.; Tao, Z.; Breger, L.; Dehay, B.; Ling, Z.; Qin, C.; Cisbani, G.; Masnata, M.; et al. Use of Adeno-Associated Virus-Mediated Delivery of Mutant Huntingtin to Study the Spreading Capacity of the Protein in Mice and Non-Human Primates. Neurobiol. Dis. 2020, 141, 104951. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Du, X.; Muramatsu, S.I.; Gomez, C.M. An MiRNA-Mediated Therapy for SCA6 Blocks IRES-Driven Translation of the CACNA1A Second Cistron. Sci. Transl. Med. 2016, 8, 347ra94. [Google Scholar] [CrossRef] [Green Version]
- Ellerby, L.M. Repeat Expansion Disorders: Mechanisms and Therapeutics. Neurotherapeutics 2019, 16, 924–927. [Google Scholar] [CrossRef] [Green Version]
- Herranz-Martin, S.; Chandran, J.; Lewis, K.; Mulcahy, P.; Higginbottom, A.; Walker, C.; Valenzuela, I.M.P.Y.; Jones, R.A.; Coldicott, I.; Iannitti, T.; et al. Viral Delivery of C9orf72 Hexanucleotide Repeat Expansions in Mice Leads to Repeat-Length-Dependent Neuropathology and Behavioural Deficits. Dis. Model. Mech. 2017, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Chew, J.; Cook, C.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Daughrity, L.M.; Castanedes-Casey, M.; Kurti, A.; Stankowski, J.N.; Disney, M.D.; et al. Aberrant Deposition of Stress Granule-Resident Proteins Linked to C9orf72-Associated TDP-43 Proteinopathy. Mol. Neurodegener. 2019, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Muntean, B.S.; Masuho, I.; Dao, M.; Sutton, L.P.; Zucca, S.; Iwamoto, H.; Patil, D.N.; Wang, D.; Birnbaumer, L.; Blakely, R.D.; et al. Gαo Is a Major Determinant of CAMP Signaling in the Pathophysiology of Movement Disorders. Cell Rep. 2021, 34, 108718. [Google Scholar] [CrossRef]
- Du, X.; Carvalho-De-Souza, J.L.; Wei, C.; Carrasquel-Ursulaez, W.; Lorenzo, Y.; Gonzalez, N.; Kubota, T.; Staisch, J.; Hain, T.; Petrossian, N.; et al. Loss-of-Function BK Channel Mutation Causes Impaired Mitochondria and Progressive Cerebellar Ataxia. Proc. Natl. Acad. Sci. USA 2020, 117, 6023–6034. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; Sasaguri, H.; Kimura, N.; Tajiri, M.; Ohkubo, T.; Ono, F.; Sakaue, F.; Kanai, K.; Hirai, T.; Sano, T.; et al. Non-Human Primate Model of Amyotrophic Lateral Sclerosis with Cytoplasmic Mislocalization of TDP-43. Brain 2012, 135, 833. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, P.A.; Bland, R.J.; Das, P.; Price, R.W.; Holloway, V.; Smithson, L.; Dicker, B.L.; During, M.J.; Young, D.; Golde, T.E. Novel Rat Alzheimer’s Disease Models Based on AAV-Mediated Gene Transfer to Selectively Increase Hippocampal Aβ Levels. Mol. Neurodegener. 2007, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theofilas, P.; Brar, S.; Stewart, K.A.; Shen, H.Y.; Sandau, U.S.; Poulsen, D.; Boison, D. Adenosine Kinase as a Target for Therapeutic Antisense Strategies in Epilepsy. Epilepsia 2011, 52, 589. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.Y.; Sun, H.; Hanthorn, M.M.; Zhi, Z.; Lan, J.Q.; Poulsen, D.J.; Wang, R.K.; Boison, D. Overexpression of Adenosine Kinase in Cortical Astrocytes and Focal Neocortical Epilepsy in Mice: Laboratory Investigation. J. Neurosurg. 2014, 120, 628–638. [Google Scholar] [CrossRef]
- Lin, L.Y.; Kelliny, S.; Liu, L.C.; Al-Hawwas, M.; Zhou, X.F.; Bobrovskaya, L. Peripheral ProBDNF Delivered by an AAV Vector to the Muscle Triggers Depression-Like Behaviours in Mice. Neurotox. Res. 2020, 38, 626–639. [Google Scholar] [CrossRef]
- Guo, H.; Deji, C.; Peng, H.; Zhang, J.; Chen, Y.; Zhang, Y.; Wang, Y. The Role of SIRT1 in the Basolateral Amygdala in Depression-like Behaviors in Mice. Genes Brain Behav. 2021, 20, e12765. [Google Scholar] [CrossRef]
- Borel, F.; Kay, M.A.; Mueller, C. Recombinant AAV as a Platform for Translating the Therapeutic Potential of RNA Interference. Mol. Ther. 2014, 22, 692–701. [Google Scholar] [CrossRef] [Green Version]
- Bofill-De Ros, X.; Gu, S. Guidelines for the Optimal Design of MiRNA-Based ShRNAs. Methods 2016, 103, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Duque, S.I.; Arnold, W.D.; Odermatt, P.; Li, X.; Porensky, P.N.; Schmelzer, L.; Meyer, K.; Kolb, S.J.; Schümperli, D.; Kaspar, B.K.; et al. A Large Animal Model of Spinal Muscular Atrophy and Correction Of. Ann. Neurol. 2015, 77, 399. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.L.; Viel, C.; Clarke, J.; Bu, J.; Chan, M.; Wang, B.; Shihabuddin, L.S.; Sardi, S.P. Viral Delivery of a MicroRNA to Gba to the Mouse Central Nervous System Models Neuronopathic Gaucher Disease. Neurobiol. Dis. 2019, 130, 104513. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Mueller, C. Design of AAV Vectors for Delivery of RNAi. Methods Mol. Biol. 2019, 1950, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Song, J.-J.; Puspita, L.; Valiulahi, P.; Shim, J.-W.; Lee, S.-H. In Vitro Generation of Mature Midbrain-Type Dopamine Neurons by Adjusting Exogenous Nurr1 and Foxa2 Expressions to Their Physiologic Patterns. Exp. Mol. Med. 2017, 49, e300. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.; Haenzi, B.; Hilton, S.; Carnicer-Lombarte, A.; Hobo, B.; Verhaagen, J.; Fawcett, J.W. Optimization of Adeno-Associated Viral Vector-Mediated Transduction of the Corticospinal Tract: Comparison of Four Promoters. Gene Ther. 2020, 28, 56–74. [Google Scholar] [CrossRef]
- Gray, S.J.; Foti, S.B.; Schwartz, J.W.; Bachaboina, L.; Taylor-Blake, B.; Coleman, J.; Ehlers, M.D.; Zylka, M.J.; McCown, T.J.; Samulski, R.J. Optimizing Promoters for Recombinant Adeno-Associated Virus-Mediated Gene Expression in the Peripheral and Central Nervous System Using Self-Complementary Vectors. Hum. Gene Ther. 2011, 22, 1143. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Matsuzaki, Y.; Konno, A.; Hirai, H. Global Knockdown of Retinoid-Related Orphan Receptor α in Mature Purkinje Cells Reveals Aberrant Cerebellar Phenotypes of Spinocerebellar Ataxia. Neuroscience 2021, 462, 328–336. [Google Scholar] [CrossRef]
- Grimm, D. The Dose Can Make the Poison: Lessons Learned from Adverse in Vivo Toxicities Caused by RNAi Overexpression. Silence 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- St Martin, J.L.; Klucken, J.; Outeiro, T.F.; Nguyen, P.; Keller-McGandy, C.; Cantuti-Castelvetri, I.; Grammatopoulos, T.N.; Standaert, D.G.; Hyman, B.T.; McLean, P.J. Dopaminergic Neuron Loss and Up-Regulation of Chaperone Protein MRNA Induced by Targeted over-Expression of Alpha-Synuclein in Mouse Substantia Nigra. J. Neurochem. 2007, 100, 1449–1457. [Google Scholar] [CrossRef]
- Hammond, S.L.; Leek, A.N.; Richman, E.H.; Tjalkens, R.B. Cellular Selectivity of AAV Serotypes for Gene Delivery in Neurons and Astrocytes by Neonatal Intracerebroventricular Injection. PLoS One 2017, 12, e0188830. [Google Scholar] [CrossRef] [Green Version]
- Kondratov, O.; Kondratova, L.; Mandel, R.J.; Coleman, K.; Savage, M.A.; Gray-Edwards, H.L.; Ness, T.J.; Rodriguez-Lebron, E.; Bell, R.D.; Rabinowitz, J.; et al. A Comprehensive Study of a 29-Capsid AAV Library in a Non-Human Primate Central Nervous System. Mol. Ther. 2021, 29, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Andres-Mateos, E.; Lerner, E.P.; Volak, A.; Cohen, O.; Hyman, B.T.; Maguire, C.A.; Vandenberghe, L.H. Efficient Gene Transfer to the Central Nervous System by Single-Stranded Anc80L65. Mol. Ther. Methods Clin. Dev. 2018, 10, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresto, N.; Gardier, C.; Gaillard, M.C.; Gubinelli, F.; Roost, P.; Molina, D.; Josephine, C.; Dufour, N.; Auregan, G.; Guillermier, M.; et al. The C-Terminal Domain of LRRK2 with the G2019S Substitution Increases Mutant A53T α-Synuclein Toxicity in Dopaminergic Neurons In Vivo. Int. J. Mol. Sci. 2021, 22, 6760. [Google Scholar] [CrossRef] [PubMed]
- Grames, M.S.; Dayton, R.D.; Jackson, K.L.; Richard, A.D.; Lu, X.; Klein, R.L. Cre-Dependent AAV Vectors for Highly Targeted Expression of Disease-Related Proteins and Neurodegeneration in the Substantia Nigra. FASEB J. 2018, 32, 4420–4427. [Google Scholar] [CrossRef] [Green Version]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic Silencing of Mutant Huntingtin with SiRNA Attenuates Striatal and Cortical Neuropathology and Behavioral Deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef] [Green Version]
- Gabery, S.; Sajjad, M.U.; Hult, S.; Soylu, R.; Kirik, D.; Petersén, Å. Characterization of a Rat Model of Huntington’s Disease Based on Targeted Expression of Mutant Huntingtin in the Forebrain Using Adeno-Associated Viral Vectors. Eur. J. Neurosci. 2012, 36, 2789–2800. [Google Scholar] [CrossRef]
- Jang, M.; Lee, S.E.; Cho, I.H. Adeno-Associated Viral Vector Serotype DJ-Mediated Overexpression of N171-82Q-Mutant Huntingtin in the Striatum of Juvenile Mice Is a New Model for Huntington’s Disease. Front. Cell. Neurosci. 2018, 12, 157. [Google Scholar] [CrossRef]
- So, K.H.; Choi, J.H.; Islam, J.; Kc, E.; Moon, H.C.; Won, S.Y.; Kim, H.K.; Kim, S.; Hyun, S.H.; Park, Y.S. An Optimization of AAV-82Q-Delivered Rat Model of Huntington’s Disease. J. Korean Neurosurg. Soc. 2020, 63, 579. [Google Scholar] [CrossRef] [Green Version]
- Audrain, M.; Fol, R.; Dutar, P.; Potier, B.; Billard, J.M.; Flament, J.; Alves, S.; Burlot, M.A.; Dufayet-Chaffaud, G.; Bemelmans, A.P.; et al. Alzheimer’s Disease-like APP Processing in Wild-Type Mice Identifies Synaptic Defects as Initial Steps of Disease Progression. Mol. Neurodegener. 2016, 11. [Google Scholar] [CrossRef]
- Jaworski, T.; Dewachter, I.; Lechat, B.; Croes, S.; Termont, A.; Demedts, D.; Borghgraef, P.; Devijver, H.; Filipkowski, R.K.; Kaczmarek, L.; et al. AAV-Tau Mediates Pyramidal Neurodegeneration by Cell-Cycle Re-Entry without Neurofibrillary Tangle Formation in Wild-Type Mice. PLoS One 2009, 4, 7280. [Google Scholar] [CrossRef]
- Jackson, K.L.; Dayton, R.D.; Deverman, B.E.; Klein, R.L. Better Targeting, Better Efficiency for Wide-Scale Neuronal Transduction with the Synapsin Promoter and AAV-PHP.B. Front. Mol. Neurosci. 2016, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Perez, B.A.; Shutterly, A.; Chan, Y.K.; Byrne, B.J.; Corti, M. Management of Neuroinflammatory Responses to AAV-Mediated Gene Therapies for Neurodegenerative Diseases. Brain Sci. 2020, 10, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehara, Y.; Fujimoto, K.I.; Ikeguchi, K.; Katakai, Y.; Ono, F.; Takino, N.; Ito, M.; Ozawa, K.; Muramatsu, S.I. Persistent Expression of Dopamine-Synthesizing Enzymes 15 Years After Gene Transfer in a Primate Model of Parkinson’s Disease. Hum. Gene Ther. Clin. Dev. 2017, 28, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Beckman, D.; Chakrabarty, P.; Ott, S.; Dao, A.; Zhou, E.; Janssen, W.G.; Donis-Cox, K.; Muller, S.; Kordower, J.H.; Morrison, J.H. A Novel Tau-based Rhesus Monkey Model of Alzheimer’s Pathogenesis. Alzheimers Dement. 2021, 17, 933. [Google Scholar] [CrossRef]
- Ulusoy, A.; Rusconi, R.; Pérez-Revuelta, B.I.; Musgrove, R.E.; Helwig, M.; Winzen-Reichert, B.; Di Monte, D.A. Caudo-Rostral Brain Spreading of α-Synuclein through Vagal Connections. EMBO Mol. Med. 2013, 5, 1051. [Google Scholar] [CrossRef]
- Jackson, K.L.; Dayton, R.D.; Klein, R.L. AAV9 Supports Wide-Scale Transduction of the CNS and TDP-43 Disease Modeling in Adult Rats. Mol. Ther. Methods Clin. Dev. 2015, 2, 15036. [Google Scholar] [CrossRef]
- Hult, S.; Soylu, R.; Björklund, T.; Belgardt, B.F.; Mauer, J.; Brüning, J.C.; Kirik, D.; Petersén, Å. Mutant Huntingtin Causes Metabolic Imbalance by Disruption of Hypothalamic Neurocircuits. Cell Metab. 2011, 13, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, I.; Fiengo, P.; Remelli, R.; Miragliotta, V.; Rossini, L.; Biotti, I.; Cappelli, A.; Petricca, L.; La Rosa, S.; Caricasole, A.; et al. Recombinant Adeno Associated Viral (AAV) Vector Type 9 Delivery of Ex1-Q138-Mutant Huntingtin in the Rat Striatum as a Short-Time Model for in Vivo Studies in Drug Discovery. Neurobiol. Dis. 2016, 86, 41–51. [Google Scholar] [CrossRef]
- Franich, N.R.; Fitzsimons, H.L.; Fong, D.M.; Klugmann, M.; During, M.J.; Young, D. AAV Vector–Mediated RNAi of Mutant Huntingtin Expression Is Neuroprotective in a Novel Genetic Rat Model of Huntington’s Disease. Mol. Ther. 2008, 16, 947–956. [Google Scholar] [CrossRef]
- Ni, S.; Huang, H.; He, D.; Chen, H.; Wang, C.; Zhao, X.; Chen, X.; Cui, W.; Zhou, W.; Zhang, J. Adeno-Associated Virus-Mediated over-Expression of CREB-Regulated Transcription Coactivator 1 in the Hippocampal Dentate Gyrus Ameliorates Lipopolysaccharide-Induced Depression-like Behaviour in Mice. J. Neurochem. 2019, 149, 111–125. [Google Scholar] [CrossRef]
- Mineur, Y.S.; Taylor, S.R.; Picciotto, M.R. Calcineurin Down-Regulation in the Amygdala Is Sufficient to Induce Anxiety-like and Depression-like Behaviors in C57BL/6J Male Mice. Biol. Psychiatry 2014, 75, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Wang, S.E.; Ko, S.Y.; Kang, H.J.; Chae, S.Y.; Lee, S.H.; Kim, Y.S.; Duman, R.S.; Son, H. Overexpression of Human GATA-1 and GATA-2 Interferes with Spine Formation and Produces Depressive Behavior in Rats. PLoS ONE 2014, 9, e109253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.; Fan, C.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Hippocampal CA1 ΒcaMKII Mediates Neuroinflammatory Responses via COX-2/PGE2 Signaling Pathways in Depression. J. Neuroinflammation 2018, 15, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased Expression of Synapse-Related Genes and Loss of Synapses in Major Depressive Disorder. Nat. Med. 2012, 18, 1413–1417. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Goedert, M. Alzheimer’s and Parkinson’s Diseases: The Prion Concept in Relation to Assembled Aβ, Tau, and α-Synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Conn, P.M. Handbook of Models for Human Aging; Elsevier Academic Press: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Platt, T.L.; Beckett, T.L.; Kohler, K.; Niedowicz, D.M.; Murphy, M.P. Obesity, Diabetes, and Leptin Resistance Promote Tau Pathology in a Mouse Model of Disease. Neuroscience 2016, 315, 162. [Google Scholar] [CrossRef] [Green Version]
- Platt, T.L.; Reeves, V.L.; Murphy, M.P. Transgenic Models of Alzheimer’s Disease: Better Utilization of Existing Models through Viral Transgenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1437–1448. [Google Scholar] [CrossRef] [Green Version]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent Models for Alzheimer Disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Ittner, L.M.; Klugmann, M.; Ke, Y.D. Adeno-associated Virus-based Alzheimer’s Disease Mouse Models and Potential New Therapeutic Avenues. Br. J. Pharmacol. 2019, 176, 3649. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Brice, A. Parkinson’s Disease: From Monogenic Forms to Genetic Susceptibility Factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Sastry, N.; Zheng, W.; Liu, G.; Wang, H.; Chen, X.; Cai, M.; Contractor, P.; Sgobio, C.; Sun, L.; Xie, C.; et al. No Apparent Transmission of Transgenic α–Synuclein into Nigrostriatal Dopaminergic Neurons in Multiple Mouse Models. Transl. Neurodegener. 2015, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, M.; Ishii, A.; Ukai, Y.; Sakagami, J.; Iwata, S.; Ono, M.; Matsumoto, K.; Nakamura, A.; Tada, N.; Kobayashi, K.; et al. Accumulation of Phosphorylated α-Synuclein in Dopaminergic Neurons of Transgenic Mice That Express Human α-Synuclein. J. Neurosci. Res. 2007, 85, 1819–1825. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Kirik, D.; Stoyka, L.E.; Standaert, D.G.; Harms, A.S. How Can RAAV-α-Synuclein and the Fibril α-Synuclein Models Advance Our Understanding of Parkinson’s Disease? J. Neurochem. 2016, 139, 131. [Google Scholar] [CrossRef]
- Albert, K.; Voutilainen, M.H.; Domanskyi, A.; Airavaara, M. AAV Vector-Mediated Gene Delivery to Substantia Nigra Dopamine Neurons: Implications for Gene Therapy and Disease Models. Genes 2017, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Recasens, A.; Ulusoy, A.; Kahle, P.J.; Di Monte, D.A.; Dehay, B. In Vivo Models of Alpha-Synuclein Transmission and Propagation. Cell Tissue Res. 2017, 373, 183–193. [Google Scholar] [CrossRef]
- Huntington, T.E.; Srinivasan, R. Adeno-Associated Virus Expression of α-Synuclein as a Tool to Model Parkinson’s Disease: Current Understanding and Knowledge Gaps. Aging Dis. 2021, 12, 1120. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Marin, B.; Hamidou, B.; Couratier, P.; Nicol, M.; Delzor, A.; Raymondeau, M.; Druet-Cabanac, M.; Lautrette, G.; Boumediene, F.; Preux, P.M. Population-Based Epidemiology of Amyotrophic Lateral Sclerosis (ALS) in an Ageing Europe – the French Register of ALS in Limousin (FRALim Register). Eur. J. Neurol. 2014, 21, 1292-e79. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and Their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 Hexanucleotide Repeat Expansion in Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: A Cross-Sectional Study. Lancet Neurol. 2012, 11, 323. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.J.W.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of Adult Huntington’s Disease in the UK Based on Diagnoses Recorded in General Practice Records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, R.A.C. Huntington’s Disease: A Clinical Review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, H.L. The Spinocerebellar Ataxias. J. Neuroophthalmol. 2009, 29, 227. [Google Scholar] [CrossRef] [Green Version]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Hache, A.; Trottier, Y. Gene Deregulation and Underlying Mechanisms in Spinocerebellar Ataxias With Polyglutamine Expansion. Front. Neurosci. 2020, 14, 571. [Google Scholar] [CrossRef]
- Ino, H. Immunohistochemical Characterization of the Orphan Nuclear Receptor RORα in the Mouse Nervous System. J. Histochem. Cytochem. 2004, 52, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Saegusa, H.; Wakamori, M.; Matsuda, Y.; Wang, J.; Mori, Y.; Zong, S.; Tanabe, T. Properties of Human Cav2.1 Channel with a Spinocerebellar Ataxia Type 6 Mutation Expressed in Purkinje Cells. Mol. Cell. Neurosci. 2007, 34, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, J.; Zhu, H.; Rinaldo, L.; Lamar, K.M.; Palmenberg, A.C.; Hansel, C.; Gomez, C.M. A Second Cistron in the CACNA1A Gene Encodes a Transcription Factor That Mediates Cerebellar Development and SCA6. Cell 2013, 154, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirnemann, J.Ô.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Vitner, E.B.; Futerman, A.H. Animal Models for Gaucher Disease Research. Dis. Model. Mech. 2011, 4, 746–752. [Google Scholar] [CrossRef] [Green Version]
- Dahl, M.; Doyle, A.; Olsson, K.; Månsson, J.E.; Marques, A.R.A.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral Gene Therapy Using Cellular Promoters Cures Type 1 Gaucher Disease in Mice. Mol. Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; De Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 4, 18024. [Google Scholar] [CrossRef]
- Ellis, C.A.; Petrovski, S.; Berkovic, S.F. Epilepsy Genetics: Clinical Impacts and Biological Insights. Lancet Neurol. 2020, 19, 93–100. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Z.J.; Liu, L.; Xu, H.Q.; Shi, Y.W.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-Associated Genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Kodera, H.; Akita, T.; Shiina, M.; Kato, M.; Hoshino, H.; Terashima, H.; Osaka, H.; Nakamura, S.; Tohyama, J.; et al. De Novo Mutations in GNAO1, Encoding a Gαo Subunit of Heterotrimeric G Proteins, Cause Epileptic Encephalopathy. Am. J. Hum. Genet. 2013, 93, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Khalil, S.; Neubig, R.R.; Sidiropoulos, C. A Mechanistic Review on GNAO1-Associated Movement Disorder. Neurobiol. Dis. 2018, 116, 131–141. [Google Scholar] [CrossRef]
- Aronica, E.; Sandau, U.S.; Iyer, A.; Boison, D. Glial Adenosine Kinase – a Neuropathological Marker of the Epileptic Brain. Neurochem. Int. 2013, 63, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, T.; Alemu Abajobir, A.; Hassen Abate, K.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Suliankatchi Abdulkader, R.; Abdulle, A.M.; Abuka Abebo, T.; Ferede Abera, S.; et al. Global, Regional, and National Incidence, Prevalence, and Years Lived with Disability for 328 Diseases and Injuries for 195 Countries, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Glob. Health Metr. 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Kessler, R.C. The Costs of Depression. Psychiatr. Clin. N. Am. 2012, 35, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitsillou, E.; Bresnehan, S.M.; Kagarakis, E.A.; Wijoyo, S.J.; Liang, J.; Hung, A.; Karagiannis, T.C. The Cellular and Molecular Basis of Major Depressive Disorder: Towards a Unified Model for Understanding Clinical Depression. Mol. Biol. Rep. 2020, 47, 753–770. [Google Scholar] [CrossRef]
- Kennis, M.; Gerritsen, L.; van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective Biomarkers of Major Depressive Disorder: A Systematic Review and Meta-Analysis. Mol. Psychiatry 2020, 25, 321–338. [Google Scholar] [CrossRef] [Green Version]
- Nestler, E.J.; Carlezon, W.A. The Mesolimbic Dopamine Reward Circuit in Depression. Biol. Psychiatry 2006, 59, 1151–1159. [Google Scholar] [CrossRef]
- Pierce, R.C.; Kumaresan, V. The Mesolimbic Dopamine System: The Final Common Pathway for the Reinforcing Effect of Drugs of Abuse? Neurosci. Biobehav. Rev. 2006, 30, 215–238. [Google Scholar] [CrossRef]
- Davis, M.; Whalen, P.J. The Amygdala: Vigilance and Emotion. Mol. Psychiatry 2000, 6, 13–34. [Google Scholar] [CrossRef] [Green Version]
- Cai, N.; Bigdeli, T.B.; Kretzschmar, W.; Lei, Y.; Liang, J.; Song, L.; Hu, J.; Li, Q.; Jin, W.; Hu, Z.; et al. Sparse Whole-Genome Sequencing Identifies Two Loci for Major Depressive Disorder. Nature 2015, 523, 588–591. [Google Scholar] [CrossRef]
- Kenworthy, C.A.; Sengupta, A.; Luz, S.M.; Ver Hoeve, E.S.; Meda, K.; Bhatnagar, S.; Abel, T. Social Defeat Induces Changes in Histone Acetylation and Expression of Histone Modifying Enzymes in the Ventral Hippocampus, Prefrontal Cortex, and Dorsal Raphe Nucleus. Neuroscience 2014, 264, 88–98. [Google Scholar] [CrossRef]
- Roddy, D.W.; Farrell, C.; Doolin, K.; Roman, E.; Tozzi, L.; Frodl, T.; O’Keane, V.; O’Hanlon, E. The Hippocampus in Depression: More Than the Sum of Its Parts? Advanced Hippocampal Substructure Segmentation in Depression. Biol. Psychiatry 2019, 85, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.H.; Son, H.; Hwang, S.; Kim, S.H. Neuropathological Abnormalities of Astrocytes, GABAergic Neurons, and Pyramidal Neurons in the Dorsolateral Prefrontal Cortices of Patients with Major Depressive Disorder. Eur. Neuropsychopharmacol. 2012, 22, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Waldsee, R.; Eftekhari, S.; Ahnstedt, H.; Johnson, L.E.; Edvinsson, L. CaMKII and MEK1/2 Inhibition Time-Dependently Modify Inflammatory Signaling in Rat Cerebral Arteries during Organ Culture. J. Neuroinflammation 2014, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- McClements, M.E.; Maclaren, R.E. Adeno-Associated Virus (AAV) Dual Vector Strategies for Gene Therapy Encoding Large Transgenes. Yale J. Biol. Med. 2017, 90, 611. [Google Scholar] [PubMed]
- Colón-Thillet, R.; Jerome, K.R.; Stone, D. Optimization of AAV Vectors to Target Persistent Viral Reservoirs. Virol. J. 2021, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.J.; Weber, W.; Fussenegger, M. Synthetic Biology: Emerging Concepts to Design and Advance Adeno-Associated Viral Vectors for Gene Therapy. Adv. Sci. 2021, 8, 2004018. [Google Scholar] [CrossRef]
- Kremer, L.P.M.; Cerrizuela, S.; Dehler, S.; Stiehl, T.; Weinmann, J.; Abendroth, H.; Kleber, S.; Laure, A.; El Andari, J.; Anders, S.; et al. High Throughput Screening of Novel AAV Capsids Identifies Variants for Transduction of Adult NSCs within the Subventricular Zone. Mol. Ther. Methods Clin. Dev. 2021, 23, 33–50. [Google Scholar] [CrossRef]
- Westhaus, A.; Cabanes-Creus, M.; Rybicki, A.; Baltazar, G.; Navarro, R.G.; Zhu, E.; Drouyer, M.; Knight, M.; Albu, R.F.; Ng, B.H.; et al. High-Throughput in Vitro, Ex Vivo, and in Vivo Screen of Adeno-Associated Virus Vectors Based on Physical and Functional Transduction. Hum. Gene Ther. 2020, 31, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.J.; Farshim, P.P.; Flomen, R.; Jones, S.T.; McAteer, S.J.; Deverman, B.E.; Gradinaru, V.; Bates, G.P. Use of High-Content Imaging to Quantify Transduction of AAV-PHP Viruses in the Brain Following Systemic Delivery. Brain Commun. 2021, 3, fcab105. [Google Scholar] [CrossRef]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Mehta, A.; Hentall, I.D.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; et al. Consequences of Zygote Injection and Germline Transfer of Mutant Human Mitochondrial DNA in Mice. Proc. Natl. Acad. Sci. USA 2015, 112, E5689–E5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunev, E.; Karan, A.; Egorova, T.; Bardina, M. Adeno-Associated Viruses for Modeling Neurological Diseases in Animals: Achievements and Prospects. Biomedicines 2022, 10, 1140. https://doi.org/10.3390/biomedicines10051140
Lunev E, Karan A, Egorova T, Bardina M. Adeno-Associated Viruses for Modeling Neurological Diseases in Animals: Achievements and Prospects. Biomedicines. 2022; 10(5):1140. https://doi.org/10.3390/biomedicines10051140
Chicago/Turabian StyleLunev, Evgenii, Anna Karan, Tatiana Egorova, and Maryana Bardina. 2022. "Adeno-Associated Viruses for Modeling Neurological Diseases in Animals: Achievements and Prospects" Biomedicines 10, no. 5: 1140. https://doi.org/10.3390/biomedicines10051140
APA StyleLunev, E., Karan, A., Egorova, T., & Bardina, M. (2022). Adeno-Associated Viruses for Modeling Neurological Diseases in Animals: Achievements and Prospects. Biomedicines, 10(5), 1140. https://doi.org/10.3390/biomedicines10051140