
Targeting of Mevalonate-Isoprenoid Pathway in Acute Myeloid Leukemia Cells by Bisphosphonate Drugs †

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
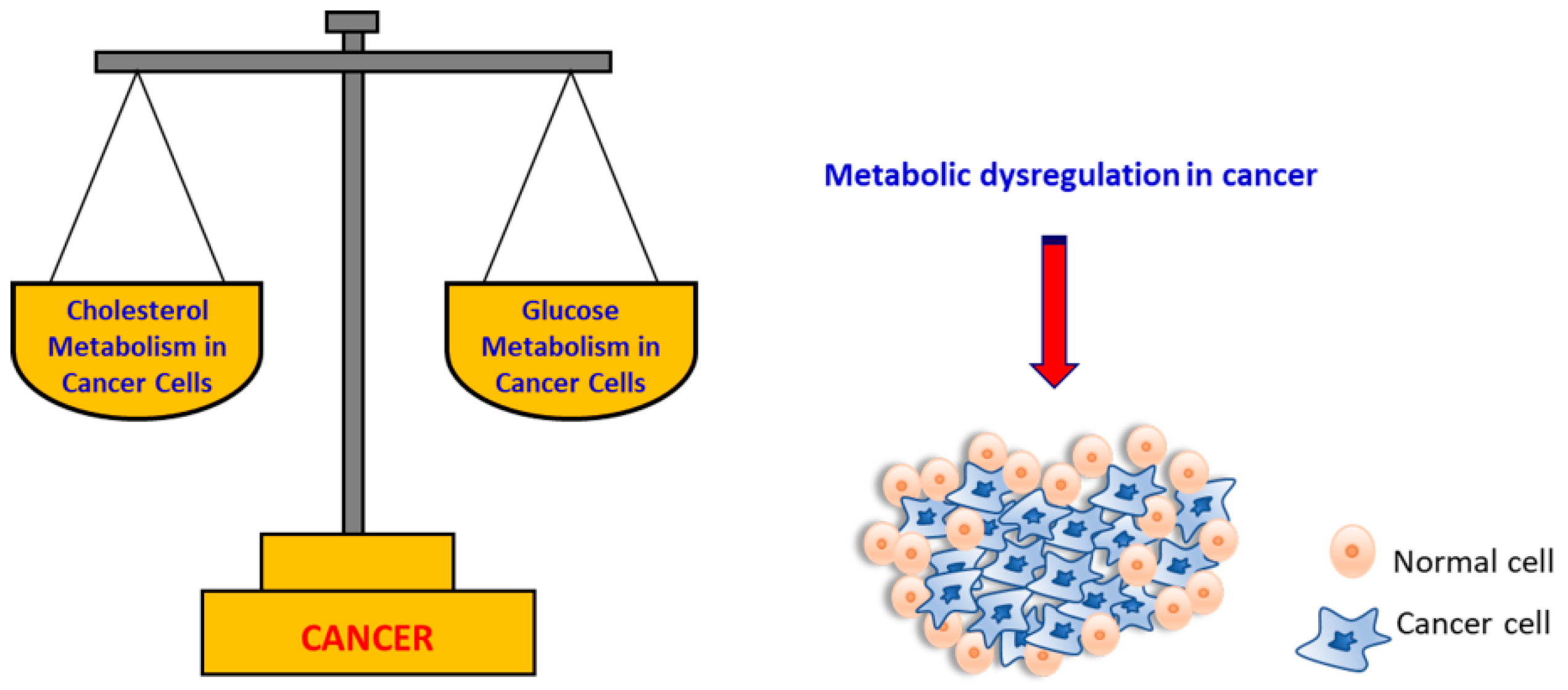
:1. Introduction
2. Biochemistry of the Mevalonate Cascade and Its Regulation in Cholesterol Metabolism
3. Regulation of Small GTPase Prenylation in AML
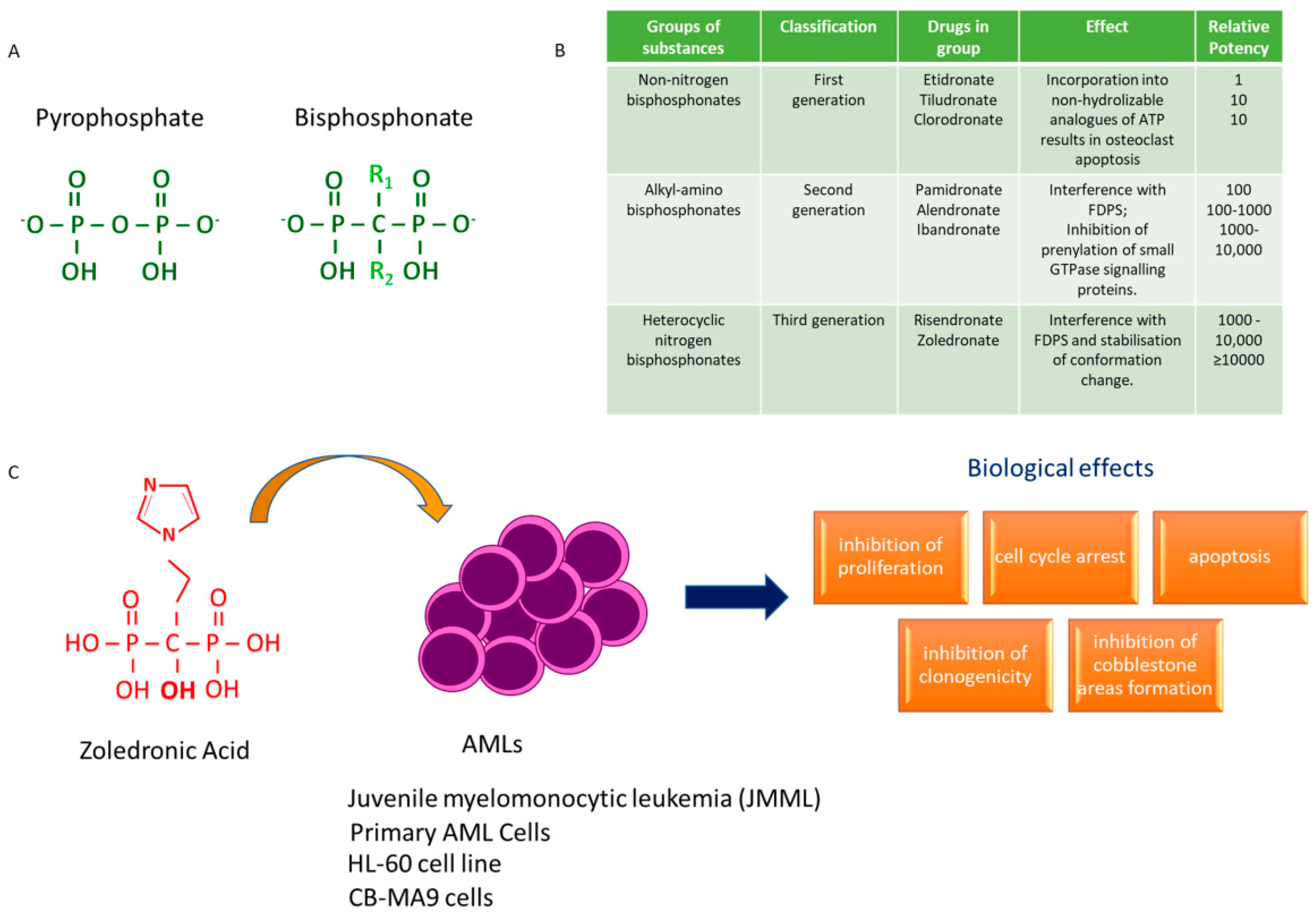
4. Bisphosphonates and AML: A Journey through Time
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kim, J.; DeBerardinis, R.J. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Schade, D.S.; Shey, L.; Eaton, R.P. Cholesterol Review: A Metabolically Important Molecule. Endocr. Pract. 2020, 26, 1514–1523. [Google Scholar] [CrossRef]
- Chiarella, E.; Lombardo, N.; Lobello, N.; Piazzetta, G.L.; Morrone, H.L.; Mesuraca, M.; Bond, H.M. Deficit in Adipose Differentiation in Mesenchymal Stem Cells Derived from Chronic Rhinosinusitis Nasal Polyps Compared to Nasal Mucosal Tissue. Int. J. Mol. Sci. 2020, 21, 9214. [Google Scholar] [CrossRef]
- Mayengbam, S.S.; Singh, A.; Pillai, A.D.; Bhat, M.K. Influence of cholesterol on cancer progression and therapy. Transl. Oncol. 2021, 14, 101043. [Google Scholar] [CrossRef]
- De Toni, L.; Sabovic, I.; De Filippis, V.; Acquasaliente, L.; Peterle, D.; Guidolin, D.; Sut, S.; Di Nisio, A.; Foresta, C.; Garolla, A. Sperm Cholesterol Content Modifies Sperm Function and TRPV1-Mediated Sperm Migration. Int. J. Mol. Sci. 2021, 22, 3126. [Google Scholar] [CrossRef]
- Aguilar-Ballester, M.; Herrero-Cervera, A.; Vinué, Á.; Martínez-Hervás, S.; González-Navarro, H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients 2020, 12, 2021. [Google Scholar] [CrossRef] [PubMed]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.L.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [Green Version]
- White, C.P. On the occurrence of crystals in tumours. J. Pathol. Bacteriol. 1909, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Robertson, T.B.; Burnett, T.C. The Influence of Lecithin and Cholesterin upon the Growth of Tumors. J. Exp. Med. 1913, 17, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M.T.; Ponce, C.; Powell, C.A.; Tarafdar, K.; Yanagita, T.; Choudhury, M.; Gollahon, L.S.; Rahman, S.M. The contribution of cholesterol and epigenetic changes to the pathophysiology of breast cancer. J. Steroid Biochem. Mol. Biol. 2018, 183, 1–9. [Google Scholar] [CrossRef]
- Revilla, G.; Pons, M.P.; Baila-Rueda, L.; García-León, A.; Santos, D.; Cenarro, A.; Magalhaes, M.; Blanco, R.M.; Moral, A.; Ignacio Pérez, J.; et al. Cholesterol and 27-hydroxycholesterol promote thyroid carcinoma aggressiveness. Sci. Rep. 2019, 9, 10260. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Jia, R. Role of de novo cholesterol synthesis enzymes in cancer. J. Cancer 2020, 11, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.F.; Zhu, N.; Zhao, T.J.; Li, H.F.; Gu, J.; Liao, D.F.; Qin, L. Involvement of LDL and ox-LDL in Cancer Development and Its Therapeutical Potential. Front. Oncol. 2022, 12, 803473. [Google Scholar] [CrossRef]
- Roslan, Z.; Muhamad, M.; Selvaratnam, L.; Ab-Rahim, S. The Roles of Low-Density Lipoprotein Receptor-Related Proteins 5, 6, and 8 in Cancer: A Review. J. Oncol. 2019, 2019, 4536302. [Google Scholar] [CrossRef] [Green Version]
- Campion, O.; Al Khalifa, T.; Langlois, B.; Thevenard-Devy, J.; Salesse, S.; Savary, K.; Schneider, C.; Etique, N.; Dedieu, S.; Devy, J. Contribution of the Low-Density Lipoprotein Receptor Family to Breast Cancer Progression. Front. Oncol. 2020, 10, 882. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, T. Cholesterol lowering: Role in cancer prevention and treatment. Biol. Chem. 2015, 396, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P., IV; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [Green Version]
- Nisticò, C.; Pagliari, F.; Chiarella, E.; Fernandes Guerreiro, J.; Marafioti, M.G.; Aversa, I.; Genard, G.; Hanley, R.; Garcia-Calderón, D.; Bond, H.M.; et al. Lipid Droplet Biosynthesis Impairment through DGAT2 Inhibition Sensitizes MCF7 Breast Cancer Cells to Radiation. Int. J. Mol. Sci. 2021, 22, 10102. [Google Scholar] [CrossRef]
- Patel, K.K.; Kashfi, K. Lipoproteins and cancer: The role of HDL-C, LDL-C, and cholesterol-lowering drugs. Biochem. Pharmacol. 2022, 196, 114654. [Google Scholar] [CrossRef]
- Schointuch, M.N.; Gilliam, T.P.; Stine, J.E.; Han, X.; Zhou, C.; Gehrig, P.A.; Kim, K.; Bae-Jump, V.L. Simvastatin, an HMG-CoA reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol. Oncol. 2014, 134, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573–588. [Google Scholar] [CrossRef]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mhaskar, R.; Kumar, A.; Miladinovic, B.; Djulbegovic, B. Bisphosphonates in multiple myeloma: An updated network meta-analysis. Cochrane Database Syst. Rev. 2017, 12, CD003188. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, S.; Zhang, Y.; Onwuka, J.U.; Zhang, Q.; Liu, X. Bisphosphonates and breast cancer survival: A meta-analysis and trial sequential analysis of 81508 participants from 23 prospective epidemiological studies. Aging 2021, 13, 19835–19866. [Google Scholar] [CrossRef]
- Zhang, G.; Gong, H.; Xu, H. Analysis of the Mechanism and Safety of Bisphosphonates in Patients with Lung Cancer and Bone Metastases. Comput. Math. Methods Med. 2021, 2021, 5343104. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ren, B.; Rao, L. Optimal duration of adjuvant bisphosphonate treatment for high-risk early breast cancer: Results from a SUCCESS trial. Thorac. Cancer 2022, 13, 519–520. [Google Scholar] [CrossRef]
- Guerra, B.; Recio, C.; Aranda-Tavío, H.; Guerra-Rodríguez, M.; García-Castellano, J.M.; Fernández-Pérez, L. The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef]
- Eckert, G.P.; Hooff, G.P.; Strandjord, D.M.; Igbavboa, U.; Volmer, D.A.; Müller, W.E.; Wood, W.G. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol. Dis. 2009, 35, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Jeong, A.; Suazo, K.F.; Wood, W.G.; Distefano, M.D.; Li, L. Isoprenoids and protein prenylation: Implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 279–310. [Google Scholar] [CrossRef]
- Cerqueira, N.M.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef]
- McClory, J.; Lin, J.T.; Timson, D.J.; Zhang, J.; Huang, M. Catalytic mechanism of mevalonate kinase revisited, a QM/MM study. Org. Biomol. Chem. 2019, 17, 2423–2431. [Google Scholar] [CrossRef]
- Anthony, J.R.; Anthony, L.C.; Nowroozi, F.; Kwon, G.; Newman, J.D.; Keasling, J.D. Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab. Eng. 2009, 11, 13–19. [Google Scholar] [CrossRef]
- Herdendorf, T.J.; Miziorko, H.M. Phosphomevalonate kinase: Functional investigation of the recombinant human enzyme. Biochemistry 2006, 45, 3235–3242. [Google Scholar] [CrossRef]
- Berthelot, K.; Estevez, Y.; Deffieux, A.; Peruch, F. Isopentenyl diphosphate isomerase: A checkpoint to isoprenoid biosynthesis. Biochimie 2012, 94, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wouters, J.; Poulter, C.D. Isopentenyl diphosphate isomerase. Mechanism-based inhibition by diene analogues of isopentenyl diphosphate and dimethylallyl diphosphate. J. Am. Chem. Soc. 2005, 127, 17433–17438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Sinensky, M. Recent advances in the study of prenylated proteins. Biochim. Biophys. Acta 2000, 1484, 93–106. [Google Scholar] [CrossRef]
- Ashok, S.; Hildebrandt, E.R.; Ruiz, C.S.; Hardgrove, D.S.; Coreno, D.W.; Schmidt, W.K.; Hougland, J.L. Protein Farnesyltransferase Catalyzes Unanticipated Farnesylation and Geranylgeranylation of Shortened Target Sequences. Biochemistry 2020, 59, 1149–1162. [Google Scholar] [CrossRef]
- Köhnke, M.; Delon, C.; Hastie, M.L.; Nguyen, U.T.; Wu, Y.W.; Waldmann, H.; Goody, R.S.; Gorman, J.J.; Alexandrov, K. Rab GTPase prenylation hierarchy and its potential role in choroideremia disease. PLoS ONE 2013, 8, e81758. [Google Scholar] [CrossRef] [Green Version]
- Jennings, B.C.; Lawton, A.J.; Rizk, Z.; Fierke, C.A. SmgGDS-607 Regulation of RhoA GTPase Prenylation Is Nucleotide-Dependent. Biochemistry 2018, 57, 4289–4298. [Google Scholar] [CrossRef]
- Padyana, A.K.; Gross, S.; Jin, L.; Cianchetta, G.; Narayanaswamy, R.; Wang, F.; Wang, R.; Fang, C.; Lv, X.; Biller, S.A.; et al. Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat. Commun. 2019, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.Y.; Shi, X.J.; Hu, A.; Wang, J.Q.; Ding, Y.; Jiang, W.; Sun, M.; Zhao, X.; Luo, J.; Qi, W.; et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature 2020, 588, 479–484. [Google Scholar] [CrossRef]
- Chen, L.; Ma, M.Y.; Sun, M.; Jiang, L.Y.; Zhao, X.T.; Fang, X.X.; Man Lam, S.; Shui, G.H.; Luo, J.; Shi, X.J.; et al. Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J. Lipid Res. 2019, 60, 1765–1775. [Google Scholar] [CrossRef]
- Pääjärvi, G.; Roudier, E.; Crisby, M.; Högberg, J.; Stenius, U. HMG-CoA reductase inhibitors, statins, induce phosphorylation of Mdm2 and attenuate the p53 response to DNA damage. FASEB J. 2005, 19, 476–478. [Google Scholar] [CrossRef]
- Clendening, J.W.; Pandyra, A.; Li, Z.; Boutros, P.C.; Martirosyan, A.; Lehner, R.; Jurisica, I.; Trudel, S.; Penn, L.Z. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood 2010, 115, 4787–4797. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhan, H.; Jiang, X.; Li, Y.; Zeng, H. The role of cholesterol metabolism in leukemia. Blood Sci. 2019, 1, 44–49. [Google Scholar] [CrossRef]
- Longo, J.; van Leeuwen, J.E.; Elbaz, M.; Branchard, E.; Penn, L.Z. Statins as Anticancer Agents in the Era of Precision Medicine. Clin. Cancer Res. 2020, 26, 5791–5800. [Google Scholar] [CrossRef]
- Park, J.; Zielinski, M.; Magder, A.; Tsantrizos, Y.S.; Berghuis, A.M. Human farnesyl pyrophosphate synthase is allosterically inhibited by its own product. Nat. Commun. 2017, 8, 14132. [Google Scholar] [CrossRef] [Green Version]
- Utriainen, P.; Niinimäki, T.T.; Huurre, A.J.; Vepsäläinen, K.L.; Mäkitie, O.M.; Niinimäki, R.A. Bisphosphonate treatment in children with acute lymphoblastic leukemia and osteonecrosis-radiological and clinical findings in a national cohort. Acta Oncol. 2021, 60, 1140–1145. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef]
- Idrees, A.S.; Sugie, T.; Inoue, C.; Murata-Hirai, K.; Okamura, H.; Morita, C.T.; Minato, N.; Toi, M.; Tanaka, Y. Comparison of γδ T cell responses and farnesyl diphosphate synthase inhibition in tumor cells pretreated with zoledronic acid. Cancer Sci. 2013, 104, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Bond, H.M.; Mesuraca, M. Regulatory Role of microRNAs Targeting the Transcription Co-Factor ZNF521 in Normal Tissues and Cancers. Int. J. Mol. Sci. 2021, 22, 8461. [Google Scholar] [CrossRef]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Todoerti, K.; Cosentino, E.G.; Lico, D.; Neri, A.; Amodio, N.; Bond, H.M.; Mesuraca, M. ZNF521 Enhances MLL-AF9-Dependent Hematopoietic Stem Cell Transformation in Acute Myeloid Leukemias by Altering the Gene Expression Landscape. Int. J. Mol. Sci. 2021, 22, 10814. [Google Scholar] [CrossRef]
- Ha, N.T.; Lee, C.H. Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells 2020, 9, 2352. [Google Scholar] [CrossRef]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef]
- Morgan, M.A.; Ganser, A.; Reuter, C.W. Therapeutic efficacy of prenylation inhibitors in the treatment of myeloid leukemia. Leukemia 2003, 17, 1482–1498. [Google Scholar] [CrossRef] [Green Version]
- Haidar, M.; Jacquemin, P. Past and Future Strategies to Inhibit Membrane Localization of the KRAS Oncogene. Int. J. Mol. Sci. 2021, 22, 13193. [Google Scholar] [CrossRef]
- Choy, E.; Chiu, V.K.; Silletti, J.; Feoktistov, M.; Morimoto, T.; Michaelson, D.; Ivanov, I.E.; Philips, M.R. Endomembrane trafficking of ras: The CAAX motif targets proteins to the ER and Golgi. Cell 1999, 98, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef]
- DerMardirossian, C.M.; Bokoch, G.M. Phosphorylation of RhoGDI by p21-activated kinase 1. Methods Enzymol. 2006, 406, 80–90. [Google Scholar] [CrossRef]
- Gray, J.L.; von Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem. Int. Ed. Engl. 2020, 59, 6342–6366. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef]
- Braun, T.; Fenaux, P. Farnesyltransferase inhibitors and their potential role in therapy for myelodysplastic syndromes and acute myeloid leukaemia. Br. J. Haematol. 2008, 141, 576–586. [Google Scholar] [CrossRef]
- Li, H.Y.; Appelbaum, F.R.; Willman, C.L.; Zager, R.A.; Banker, D.E. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood 2003, 101, 3628–3634. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Dai, X.; Wang, Y. 5-Aza-2′-deoxycytidine induced growth inhibition of leukemia cells through modulating endogenous cholesterol biosynthesis. Mol. Cell Proteom. 2012, 11, M111.016915. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J. Farnesyltransferase inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Clin. Lymphoma 2003, 4, S30–S35. [Google Scholar] [CrossRef]
- Morgan, M.; Wegner, J.; Aydilek, E.; Ganser, A.; Reuter, C.W. Synergistic cytotoxic effects in myeloid leukemia cells upon cotreatment with farnesyltransferase and geranylgeranyl transferase-I inhibitors. Leukemia 2003, 17, 1508–1520. [Google Scholar] [CrossRef]
- Rogers, M.J.; Munoz, M.A. From vesicle to cytosol. eLife 2018, 7, e38847. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Georgiou, G.; Benetatos, L.; Briasoulis, E. Gene mutations and molecularly targeted therapies in acute myeloid leukemia. Am. J. Blood Res. 2013, 3, 29–51. [Google Scholar]
- Nardone, V.; D’Asta, F.; Brandi, M.L. Pharmacological management of osteogenesis. Clinics 2014, 69, 438–446. [Google Scholar] [CrossRef]
- Santini, D.; Fratto, M.E.; Vincenzi, B.; La Cesa, A.; Dianzani, C.; Tonini, G. Bisphosphonate effects in cancer and inflammatory diseases: In vitro and in vivo modulation of cytokine activities. BioDrugs 2004, 18, 269–278. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Bisphosphonates for the treatment of osteoporosis: Insights for clinicians. Ther. Adv. Chronic Dis. 2010, 1, 115–128. [Google Scholar] [CrossRef]
- Kuźnik, A.; Październiok-Holewa, A.; Jewula, P.; Kuźnik, N. Bisphosphonates-much more than only drugs for bone diseases. Eur. J. Pharmacol. 2020, 866, 172773. [Google Scholar] [CrossRef]
- Ukon, Y.; Makino, T.; Kodama, J.; Tsukazaki, H.; Tateiwa, D.; Yoshikawa, H.; Kaito, T. Molecular-Based Treatment Strategies for Osteoporosis: A Literature Review. Int. J. Mol. Sci. 2019, 20, 2557. [Google Scholar] [CrossRef] [Green Version]
- Giudice, A.; Antonelli, A.; Chiarella, E.; Baudi, F.; Barni, T.; Di Vito, A. The Case of Medication-Related Osteonecrosis of the Jaw Addressed from a Pathogenic Point of View. Innovative Therapeutic Strategies: Focus on the Most Recent Discoveries on Oral Mesenchymal Stem Cell-Derived Exosomes. Pharmaceuticals 2020, 13, 423. [Google Scholar] [CrossRef]
- Grey, A.; Reid, I.R. Differences between the bisphosphonates for the prevention and treatment of osteoporosis. Ther. Clin. Risk Manag. 2006, 2, 77–86. [Google Scholar] [PubMed]
- Ebetino, F.H.; Sun, S.; Cherian, P.; Roshandel, S.; Neighbors, J.D.; Hu, E.; Dunford, J.E.; Sedghizadeh, P.P.; McKenna, C.E.; Srinivasan, V.; et al. Bisphosphonates: The role of chemistry in understanding their biological actions and structure-activity relationships, and new directions for their therapeutic use. Bone 2020, 156, 116289. [Google Scholar] [CrossRef]
- Rogers, M.J.; Mönkkönen, J.; Munoz, M.A. Molecular mechanisms of action of bisphosphonates and new insights into their effects outside the skeleton. Bone 2020, 139, 115493. [Google Scholar] [CrossRef]
- Jiang, L.; Cui, X.; Ma, H.; Tang, X. Comparison of denosumab and zoledronic acid for the treatment of solid tumors and multiple myeloma with bone metastasis: A systematic review and meta-analysis based on randomized controlled trials. J. Orthop. Surg. Res. 2021, 16, 400. [Google Scholar] [CrossRef] [PubMed]
- Akoury, E.; Ahangar, P.; Nour, A.; Lapointe, J.; Guérard, K.P.; Haglund, L.; Rosenzweig, D.H.; Weber, M.H. Low-dose zoledronate for the treatment of bone metastasis secondary to prostate cancer. Cancer Cell Int. 2019, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Di Vito, A.; Chiarella, E.; Baudi, F.; Scardamaglia, P.; Antonelli, A.; Giudice, D.; Barni, T.; Fortunato, L.; Giudice, A. Dose-Dependent Effects of Zoledronic Acid on Human Periodontal Ligament Stem Cells: An In Vitro Pilot Study. Cell Transplant. 2020, 29, 963689720948497. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Thompson, K.; Gordon, S.; Rogers, M.J. Molecular mechanisms of action of bisphosphonates: Current status. Clin. Cancer Res. 2006, 12, 6222s–6230s. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, J.; Kimura, S.; Segawa, H.; Kobayashi, Y.; Yoshikawa, T.; Urasaki, Y.; Ueda, T.; Enjo, F.; Tokuda, H.; Ottmann, O.G.; et al. The third-generation bisphosphonate Zoledronic Acid synergistically augments the anti-Ph+ leukemia activity of imatinib mesylate. Blood 2003, 102, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Chuah, C.; Barnes, D.J.; Kwok, M.; Corbin, A.; Deininger, M.W.; Druker, B.J.; Melo, J.V. Zoledronate inhibits proliferation and induces apoptosis of imatinib-resistant chronic myeloid leukaemia cells. Leukemia 2005, 19, 1896–1904. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuka, Y.; Manabe, A.; Kawasaki, H.; Hasegawa, D.; Zaike, Y.; Watanabe, S.; Tanizawa, T.; Nakahata, T.; Tsuji, K. RAS-blocking bisphosphonate zoledronic acid inhibits the abnormal proliferation and differentiation of juvenile myelomonocytic leukemia cells in vitro. Blood 2005, 106, 3134–3141. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, S.; Divine, C.L.; Aljitawi, O.S.; Abhyankar, S.; McGuirk, J.P.; Graves, L. Prophylactic use of zoledronic acid to prevent early bone loss is safe and feasible in patients with acute myeloid leukemia undergoing allogeneic stem cell transplantation. Clin. Transplant. 2012, 26, 447–453. [Google Scholar] [CrossRef]
- Lee, C.Y.; Suzuki, J.B. Medication-related osteonecrosis of the jaws from once per year intravenous zoledronic acid (Reclast): Report of 4 cases. Implant Dent. 2015, 24, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Kunzmann, V.; Wilhelm, M. Adjuvant zoledronic acid for breast cancer: Mechanism of action? Lancet Oncol. 2011, 12, 991–992. [Google Scholar] [CrossRef]
- Gober, H.J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Benzaïd, I.; Mönkkönen, H.; Stresing, V.; Bonnelye, E.; Green, J.; Mönkkönen, J.; Touraine, J.L.; Clézardin, P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res. 2011, 71, 4562–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundermann, S.; Klinker, E.; Kimmel, B.; Flierl, U.; Wilhelm, M.; Einsele, H.; Kunzmann, V. comprehensive analysis of primary acute myeloid leukemia identifies biomarkers predicting susceptibility to human allogeneic Vγ9Vδ2 T cells. J. Immunother. 2014, 37, 321–330. [Google Scholar] [CrossRef]
- Fan, R.F.; Chen, Y.X.; Fang, Z.G.; Guo, X.Y.; Lu, Y.; Liu, L.L.; Xu, Y.C.; Liu, X.F.; Lin, D.J. Zoledronic acid overcomes adriamycin resistance in acute myeloid leukemia cells by promoting apoptosis. Mol. Med. Rep. 2016, 14, 5660–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesuraca, M.; Amodio, N.; Chiarella, E.; Scicchitano, S.; Aloisio, A.; Codispoti, B.; Lucchino, V.; Montalcini, Y.; Bond, H.M.; Morrone, G. Turning Stem Cells Bad: Generation of Clinically Relevant Models of Human Acute Myeloid Leukemia through Gene Delivery- or Genome Editing-Based Approaches. Molecules 2018, 23, 2060. [Google Scholar] [CrossRef] [Green Version]
- Codispoti, B.; Rinaldo, N.; Chiarella, E.; Lupia, M.; Spoleti, C.B.; Marafioti, M.G.; Aloisio, A.; Scicchitano, S.; Giordano, M.; Nappo, G.; et al. Recombinant TAT-BMI-1 fusion protein induces ex vivo expansion of human umbilical cord blood-derived hematopoietic stem cells. Oncotarget 2017, 8, 43782–43798. [Google Scholar] [CrossRef] [Green Version]
- Chiarella, E.; Codispoti, B.; Aloisio, A.; Cosentino, E.G.; Scicchitano, S.; Montalcini, Y.; Lico, D.; Morrone, G.; Mesuraca, M.; Bond, H.M. Zoledronic acid inhibits the growth of leukemic MLL-AF9 transformed hematopoietic cells. Heliyon 2020, 6, e04020. [Google Scholar] [CrossRef]
- Caraglia, M.; Marra, M.; Leonetti, C.; Meo, G.; D’Alessandro, A.M.; Baldi, A.; Santini, D.; Tonini, G.; Bertieri, R.; Zupi, G.; et al. R115777 (Zarnestra)/Zoledronic acid (Zometa) cooperation on inhibition of prostate cancer proliferation is paralleled by Erk/Akt inactivation and reduced Bcl-2 and bad phosphorylation. J. Cell. Physiol. 2007, 211, 533–543. [Google Scholar] [CrossRef]
- Surmeli, Z.; Gursoy, P.; Erdogan, A.P.; Bozkurt, E.; Atmaca, H.; Uzunoglu, S.; Sezgin, C.; Şanlı, U.A.; Uslu, R.; Karaca, B. Combination of zoledronic acid and serine/threonine phosphatase inhibitors induces synergistic cytotoxicity and apoptosis in human breast cancer cells via inhibition of PI3K/Akt pathway. Tumour Biol. 2016, 37, 3665–3673. [Google Scholar] [CrossRef]
- Iizuka-Ohashi, M.; Watanabe, M.; Sukeno, M.; Morita, M.; Hoang, N.; Kuchimaru, T.; Kizaka-Kondoh, S.; Sowa, Y.; Sakaguchi, K.; Taguchi, T.; et al. Blockage of the mevalonate pathway overcomes the apoptotic resistance to MEK inhibitors with suppressing the activation of Akt in cancer cells. Oncotarget 2018, 9, 19597–19612. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Xia, H.; Zhou, S.; Tang, Q.; Bi, F. Zoledronic acid enhances the efficacy of the MEK inhibitor trametinib in KRAS mutant cancers. Cancer Lett. 2019, 442, 202–212. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarella, E.; Nisticò, C.; Di Vito, A.; Morrone, H.L.; Mesuraca, M. Targeting of Mevalonate-Isoprenoid Pathway in Acute Myeloid Leukemia Cells by Bisphosphonate Drugs. Biomedicines 2022, 10, 1146. https://doi.org/10.3390/biomedicines10051146
Chiarella E, Nisticò C, Di Vito A, Morrone HL, Mesuraca M. Targeting of Mevalonate-Isoprenoid Pathway in Acute Myeloid Leukemia Cells by Bisphosphonate Drugs. Biomedicines. 2022; 10(5):1146. https://doi.org/10.3390/biomedicines10051146
Chicago/Turabian StyleChiarella, Emanuela, Clelia Nisticò, Anna Di Vito, Helen Linda Morrone, and Maria Mesuraca. 2022. "Targeting of Mevalonate-Isoprenoid Pathway in Acute Myeloid Leukemia Cells by Bisphosphonate Drugs" Biomedicines 10, no. 5: 1146. https://doi.org/10.3390/biomedicines10051146