
Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice

 , , , , , , and
, , , , , , and 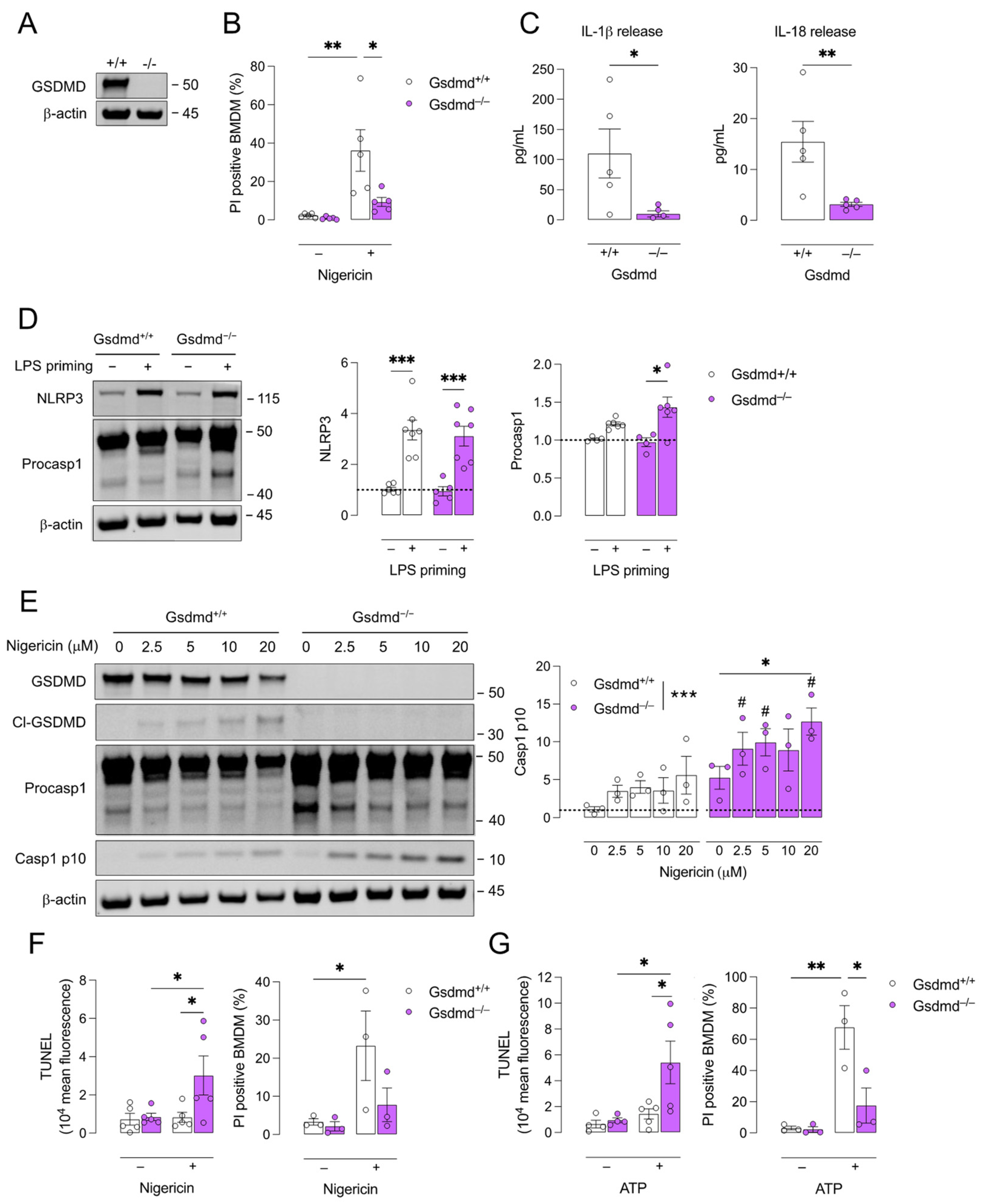{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Atherosclerotic Plaques
2.2. Mice
2.3. Histological Analyses
2.4. Cell Culture
2.5. Flow Cytometry
2.6. Western Blotting
2.7. Statistical Analyses
3. Results
3.1. Pyroptosis Is Inhibited in Gsdmd−/− BMDMs but a Switch to Apoptosis Is Induced
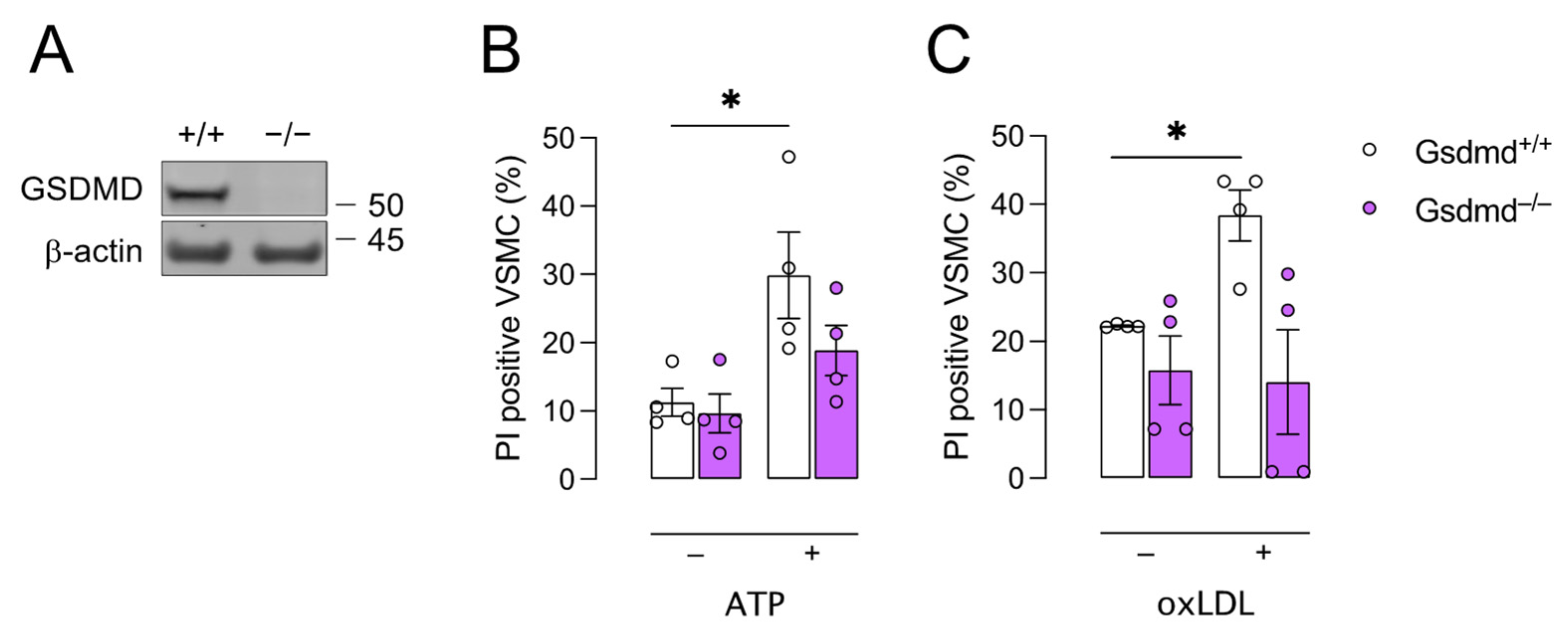
3.2. Gsdmd−/− VSMCs Are Less Sensitive to Pyroptosis Inducers
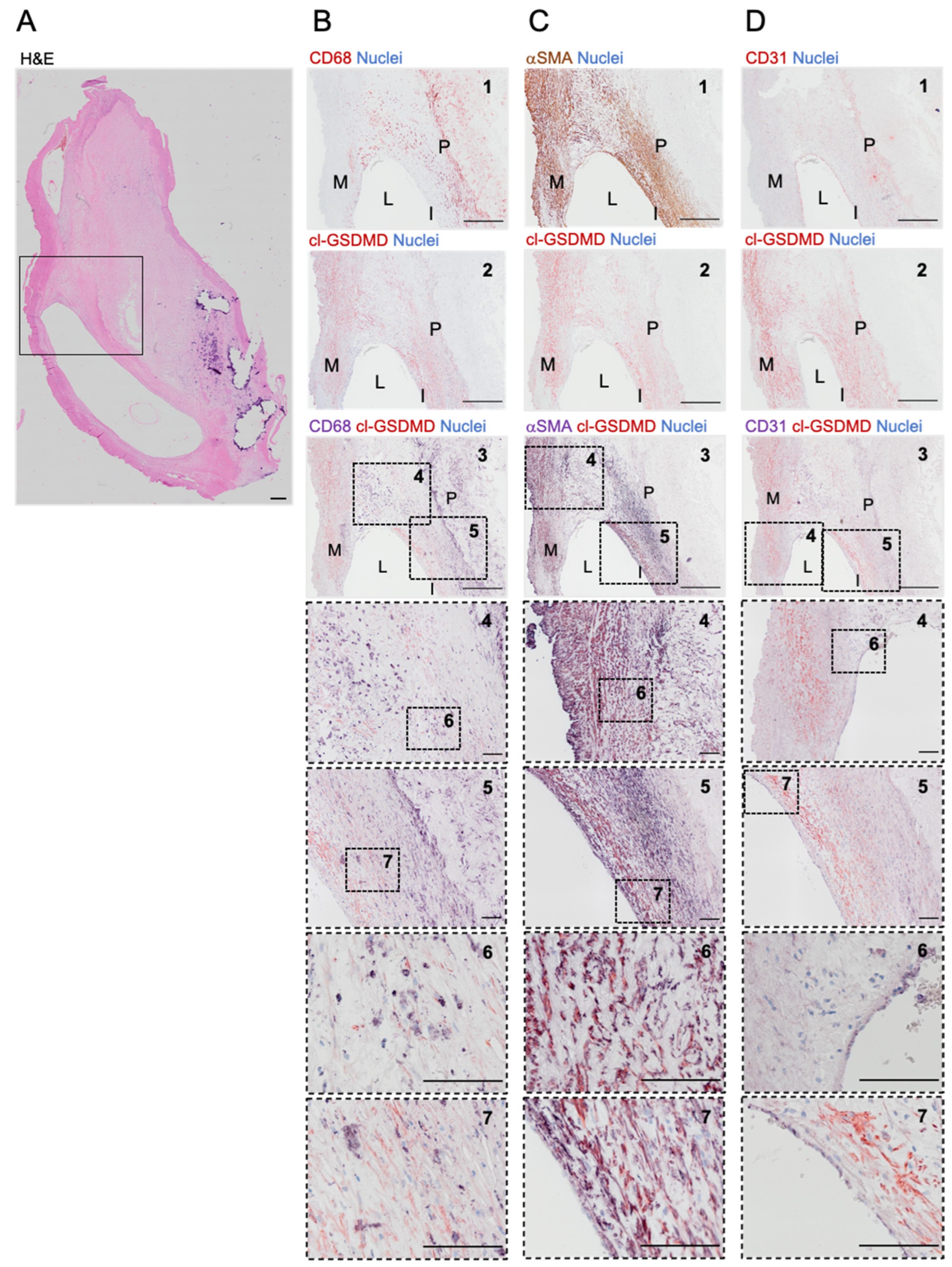
3.3. Cleaved GSDMD Is Expressed in Human Carotid Lesions
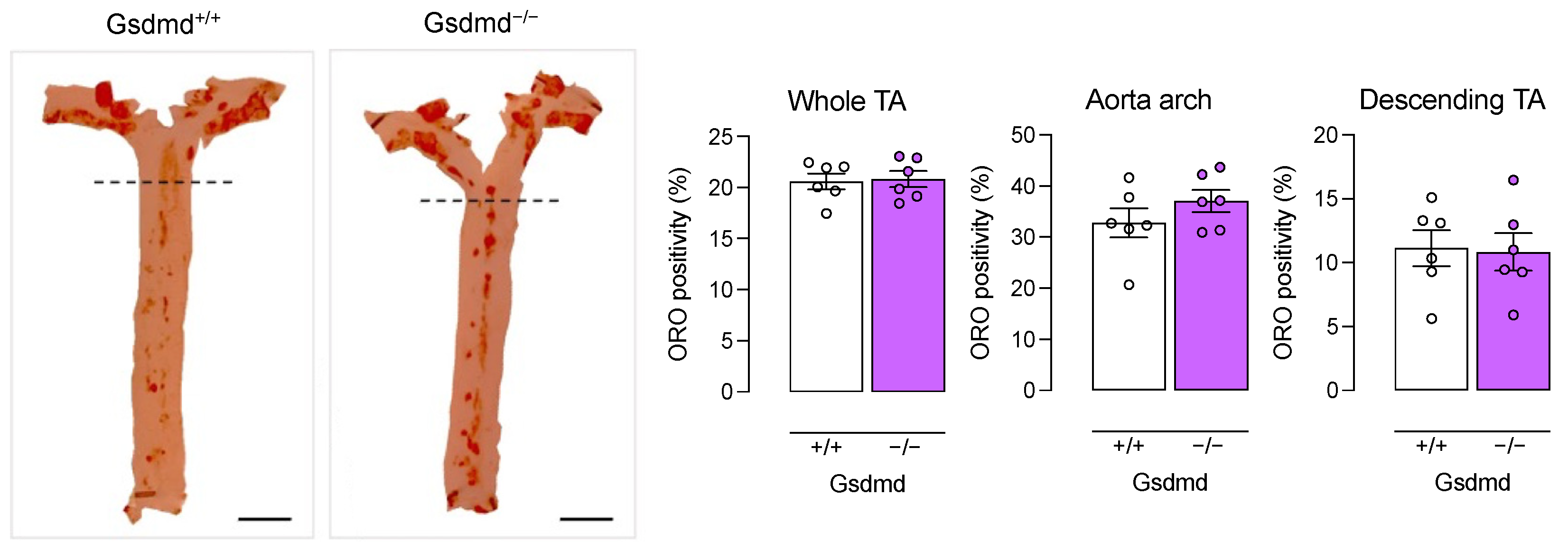
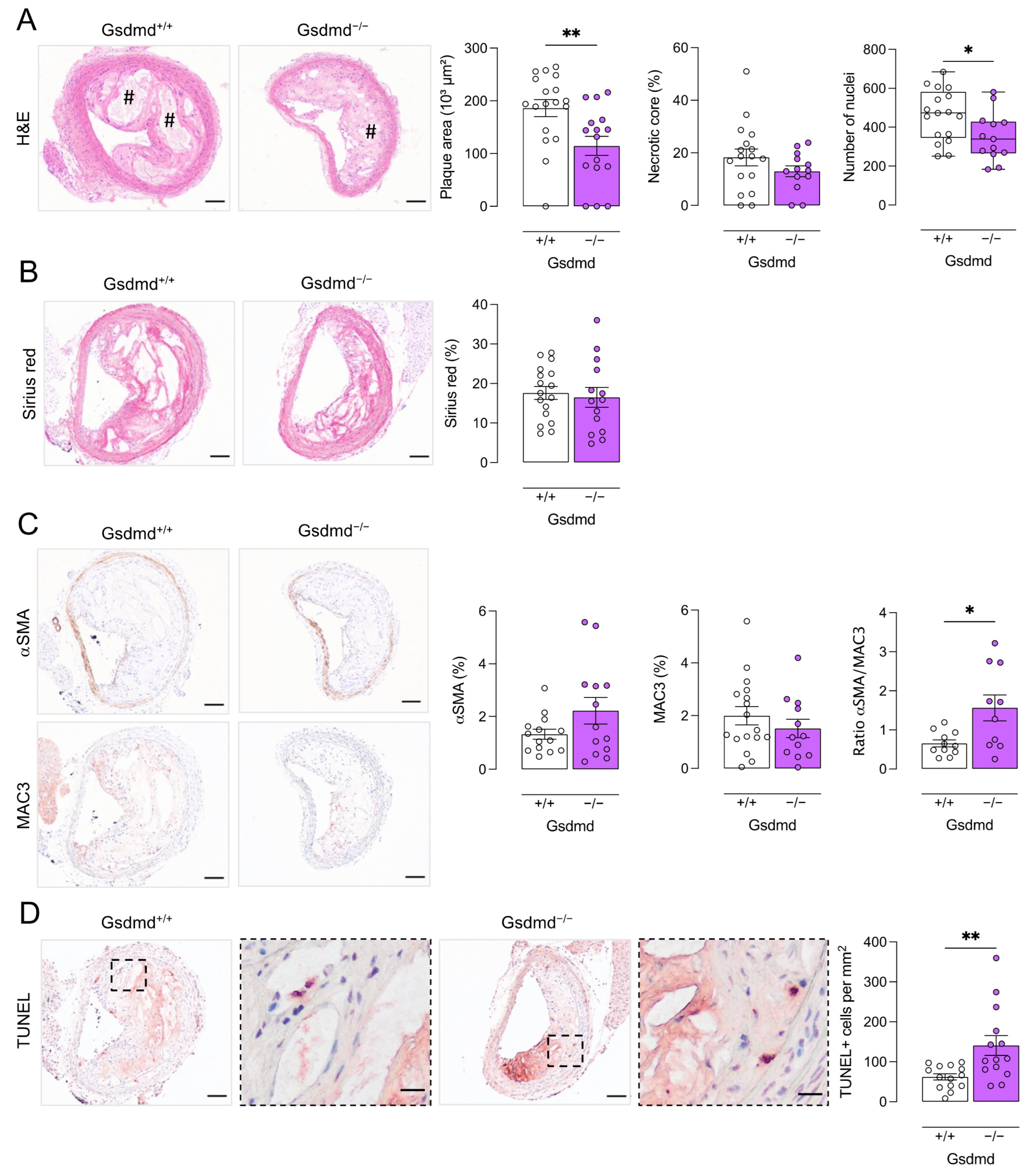
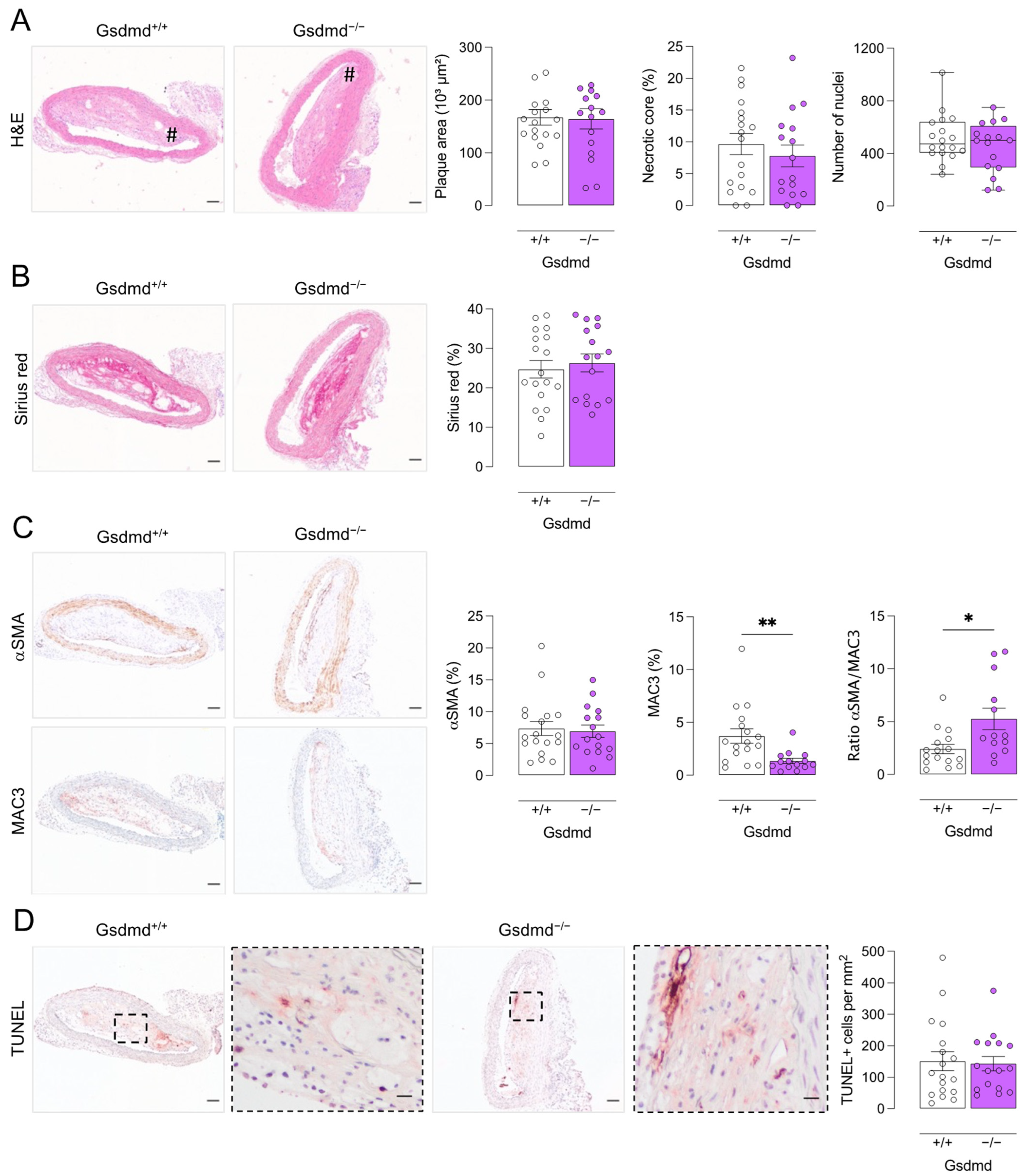
3.4. Atherogenesis Is Delayed in ApoE−/− Gsdmd−/− Mice but a Switch to Apoptosis Occurs in Plaques of the Brachiocephalic Artery
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Chen, Y.; Sun, Y.; Gao, Q.; Yu, B.; Jiang, X.; Guo, M. Gasdermin family: A promising therapeutic target for cancers and inflammation-driven diseases. J. Cell Commun. Signal. 2020, 14, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Rajamäki, K.; Lappalainen, J.; Oörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef] [Green Version]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Front. Cell Dev. Biol. 2020, 8, 167. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. The crystal-induced activation of NLRP3 inflammasomes in atherosclerosis. Inflamm. Regen. 2017, 37, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Shou, X.; Mao, X.; Dong, J.; Mohabeer, N.; Kushwaha, K.K.; Wang, L.; Su, Y.; Fang, H.; Li, D. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: Implication in lesion instability? PLoS ONE 2013, 8, e62148. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sorensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Lesèche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamaki, K.; Mayranpaa, M.I.; Risco, A.; Tuimala, J.; Nurmi, K.; Cuenda, A.; Eklund, K.K.; Oorni, K.; Kovanen, P.T. p38delta MAPK: A Novel Regulator of NLRP3 Inflammasome Activation With Increased Expression in Coronary Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1937–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Narula, J.; Burke, A.P.; Haider, N.; Farb, A.; Hui-Liang, Y.; Smialek, J.; Virmani, R. Localization of Apoptotic Macrophages at the Site of Plaque Rupture in Sudden Coronary Death. Am. J. Pathol. 2000, 157, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol. 2012, 28, 222–229. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Niu, X.; Xu, H.; Li, Q.; Meng, L.; He, M.; Zhang, J.; Zhang, Z.; Zhang, Z. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp. Cell Res. 2020, 389, 111847. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ji, N.; Gong, X.; Ni, S.; Xu, L.; Zhang, H. Thioredoxin-1 attenuates atherosclerosis development through inhibiting NLRP3 inflammasome. Endocrine 2020, 70, 65–70. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Y.; Xiong, G.; Whyte, S.; Duan, J.; Yang, Y.; Wang, K.; Yang, S.; Geng, Y.; Ou, Y.; et al. Caspase-11, a specific sensor for intracellular lipopolysaccharide recognition, mediates the non-canonical inflammatory pathway of pyroptosis. Cell Biosci. 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Sun, X.; Liu, S.; Tang, Y.; Shi, Y.; Bai, Y.; Wang, Y.; Yang, Q.; Yang, Q.; Jiang, W.; et al. Caspase-11-Gasdermin D-Mediated Pyroptosis Is Involved in the Pathogenesis of Atherosclerosis. Front. Pharmacol. 2021, 12, 657486. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Tu, Z.; Chen, K.; Xu, Y.; Chen, F.; Xu, S.; Shi, T.; Qian, J.; Shen, L.; Hwa, J.; et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J. Clin. Investig. 2022, 132, e151268. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.M.; De Meyer, G.R.; Muhring, J.; Jacob, W.; Bult, H.; Herman, A.G. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation 1998, 97, 2307–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurdi, A.; Roth, L.; Van der Veken, B.; Van Dam, D.; De Deyn, P.P.; De Doncker, M.; Neels, H.; De Meyer, G.R.Y.; Martinet, W. Everolimus depletes plaque macrophages, abolishes intraplaque neovascularization and improves survival in mice with advanced atherosclerosis. Vasc. Pharmacol. 2019, 113, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K.; Loeb, A.; Gordon, D.; Thompson, M.M. Expression of smooth muscle-specific alpha-isoactin in cultured vascular smooth muscle cells: Relationship between growth and cytodifferentiation. J. Cell Biol. 1986, 102, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Syed, F.M.; Hahn, H.S.; Odley, A.; Guo, Y.; Vallejo, J.G.; Lynch, R.A.; Mann, D.L.; Bolli, R.; Dorn, G.W., 2nd. Proapoptotic effects of caspase-1/interleukin-converting enzyme dominate in myocardial ischemia. Circ. Res. 2005, 96, 1103–1109. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, M.; Peters, A.; Becker, A.; Böckler, D.; Dihlmann, S. Inflammation-related induction of absent in melanoma 2 (AIM2) in vascular cells and atherosclerotic lesions suggests a role in vascular pathogenesis. J. Vasc. Surg. 2014, 59, 794–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef]
- Xu, Y.J.; Zheng, L.; Hu, Y.W.; Wang, Q. Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 2018, 476, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Zhaolin, Z.; Jiaojiao, C.; Peng, W.; Yami, L.; Tingting, Z.; Jun, T.; Shiyuan, W.; Jinyan, X.; Dangheng, W.; Zhisheng, J.; et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J. Cell Physiol. 2019, 234, 7475–7491. [Google Scholar] [CrossRef] [PubMed]
- Opoku, E.; Traughber, C.A.; Zhang, D.; Iacano, A.J.; Khan, M.; Han, J.; Smith, J.D.; Gulshan, K. Gasdermin D mediates inflammation-induced defects in reverse cholesterol transport and promotes atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 715211. [Google Scholar] [CrossRef]
- Lopez-Pastrana, J.; Ferrer, L.M.; Li, Y.F.; Xiong, X.; Xi, H.; Cueto, R.; Nelson, J.; Sha, X.; Li, X.; Cannella, A.L.; et al. Inhibition of Caspase-1 Activation in Endothelial Cells Improves Angiogenesis: A Novel Therapeutic Potential For Ischemia. J. Biol. Chem. 2015, 290, 17485–17494. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Yang, Y.; Ji, S.; Wang, S.; Peng, X.; Tian, C.; Sun, R.C.; Yu, T.; Chu, X.M. MicroRNA-302c-3p inhibits endothelial cell pyroptosis via directly targeting NOD-, LRR- and pyrin domain-containing protein 3 in atherosclerosis. J. Cell Mol. Med. 2021, 25, 4373–4386. [Google Scholar] [CrossRef]
- Xing, S.S.; Yang, J.; Li, W.J.; Li, J.; Chen, L.; Yang, Y.T.; Lei, X.; Li, J.; Wang, K.; Liu, X. Salidroside Decreases Atherosclerosis Plaque Formation via Inhibiting Endothelial Cell Pyroptosis. Inflammation 2020, 43, 433–440. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. 1994, 14, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.; Johnson, J.L.; Carson, K.G.; Jackson, C.L. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengrenyuk, Y.; Kaplan, T.J.; Cardoso, L.; Randolph, G.J.; Weinbaum, S. Computational stress analysis of atherosclerotic plaques in ApoE knockout mice. Ann. Biomed. Eng. 2010, 38, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.E.; Polinsky, P.; Virmani, R.; Kauser, K.; Rubanyi, G.; Schwartz, S.M. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2587–2592. [Google Scholar] [CrossRef] [Green Version]
- Meir, K.S.; Leitersdorf, E. Atherosclerosis in the apolipoprotein-E-deficient mouse: A decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Charriaut-Marlangue, C.; Ben-Ari, Y. A cautionary note on the use of the TUNEL stain to determine apoptosis. Neuroreport 1995, 7, 61–64. [Google Scholar] [CrossRef]
- Grasl-Kraupp, B.; Ruttkay-Nedecky, B.; Koudelka, H.; Bukowska, K.; Bursch, W.; Schulte-Hermann, R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: A cautionary note. Hepatology 1995, 21, 1465–1468. [Google Scholar] [CrossRef]
- Coornaert, I.; Puylaert, P.; Marcasolli, G.; Grootaert, M.O.J.; Vandenabeele, P.; De Meyer, G.R.Y.; Martinet, W. Impact of myeloid RIPK1 gene deletion on atherogenesis in ApoE-deficient mice. Atherosclerosis 2021, 322, 51–60. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puylaert, P.; Van Praet, M.; Vaes, F.; Neutel, C.H.G.; Roth, L.; Guns, P.-J.; De Meyer, G.R.Y.; Martinet, W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice. Biomedicines 2022, 10, 1171. https://doi.org/10.3390/biomedicines10051171
Puylaert P, Van Praet M, Vaes F, Neutel CHG, Roth L, Guns P-J, De Meyer GRY, Martinet W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice. Biomedicines. 2022; 10(5):1171. https://doi.org/10.3390/biomedicines10051171
Chicago/Turabian StylePuylaert, Pauline, Melissa Van Praet, Frederik Vaes, Cédric H. G. Neutel, Lynn Roth, Pieter-Jan Guns, Guido R. Y. De Meyer, and Wim Martinet. 2022. "Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice" Biomedicines 10, no. 5: 1171. https://doi.org/10.3390/biomedicines10051171
APA StylePuylaert, P., Van Praet, M., Vaes, F., Neutel, C. H. G., Roth, L., Guns, P.-J., De Meyer, G. R. Y., & Martinet, W. (2022). Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in ApoE Knock-Out Mice. Biomedicines, 10(5), 1171. https://doi.org/10.3390/biomedicines10051171