
Analysis of Mitochondrial Function in Cell Membranes as Indicator of Tissue Vulnerability to Drugs in Humans

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Tissue Samples
2.3. Cell Membrane Microarray Fabrication
2.4. Determination of Drug Effects on Superoxide Formation Promoted by NADH in Isolated Bovine Heart Cell Membranes
2.5. Determination of Tissue-Specific Effects of Drugs on Superoxide Formation Triggered by NADH Using Human CMMAs
2.6. Determination of Tissue-Specific Effect on Superoxide Formation Induced by Succinate Using Human CMMAs
2.7. Determination of Drug Tissue-Specific Effect on Cytochrome C Oxidase Activity
2.8. Data Analysis and Normalization
3. Results
3.1. Protocol Optimization of Superoxide Formation in Bovine Heart Membranes Homogenates
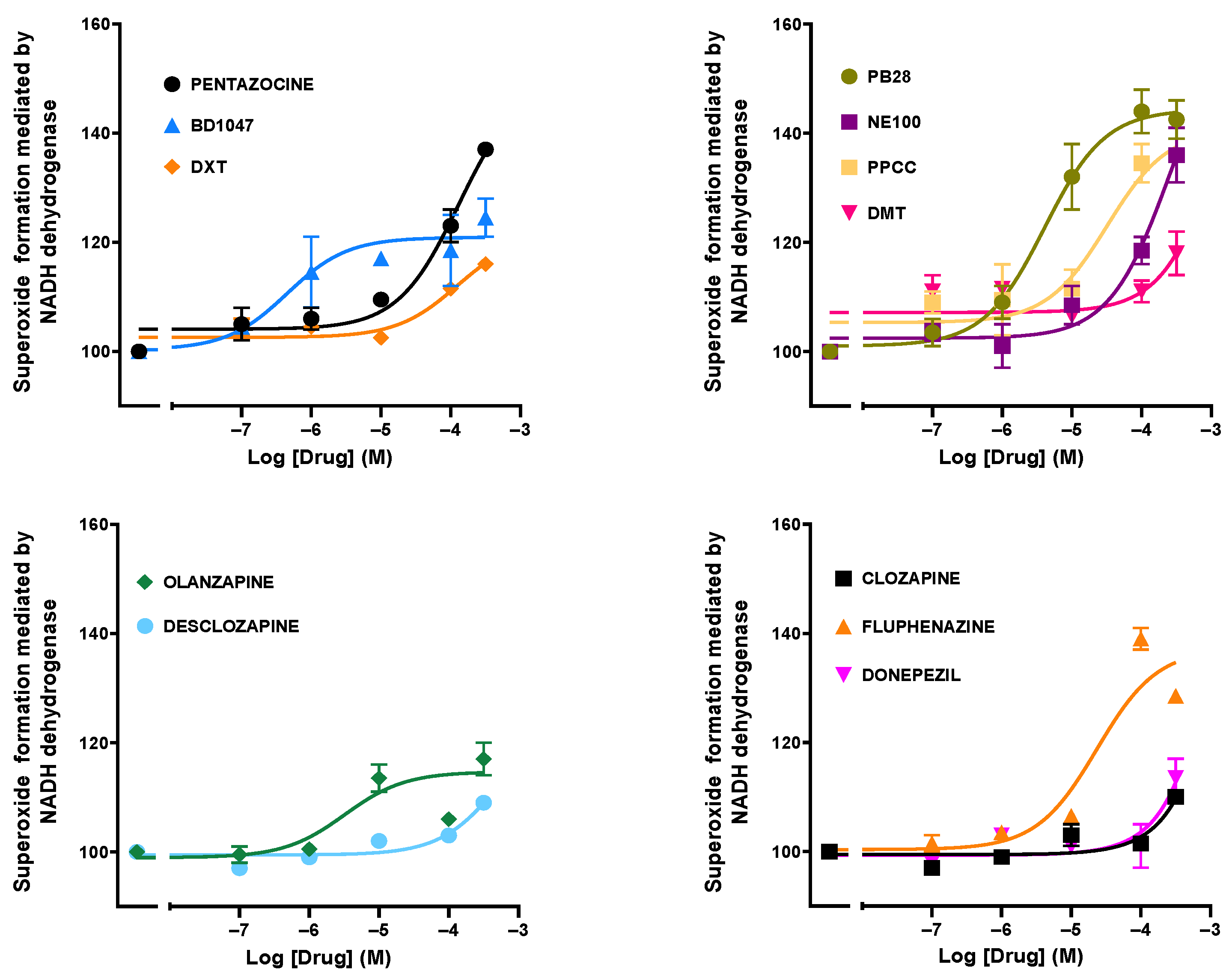
3.2. Effect of Drugs and Compounds on Superoxide Formation in Isolated Bovine Heart Membranes
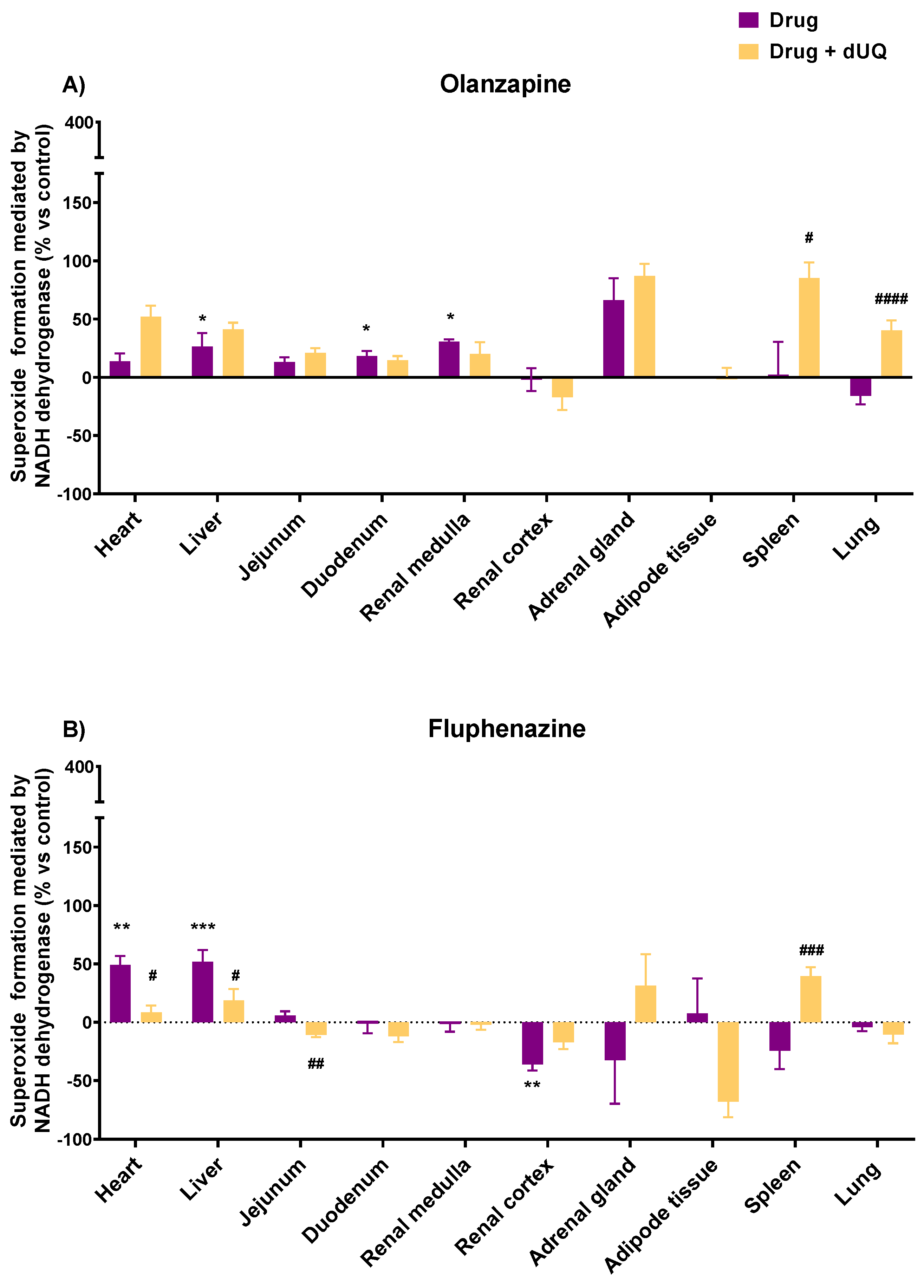
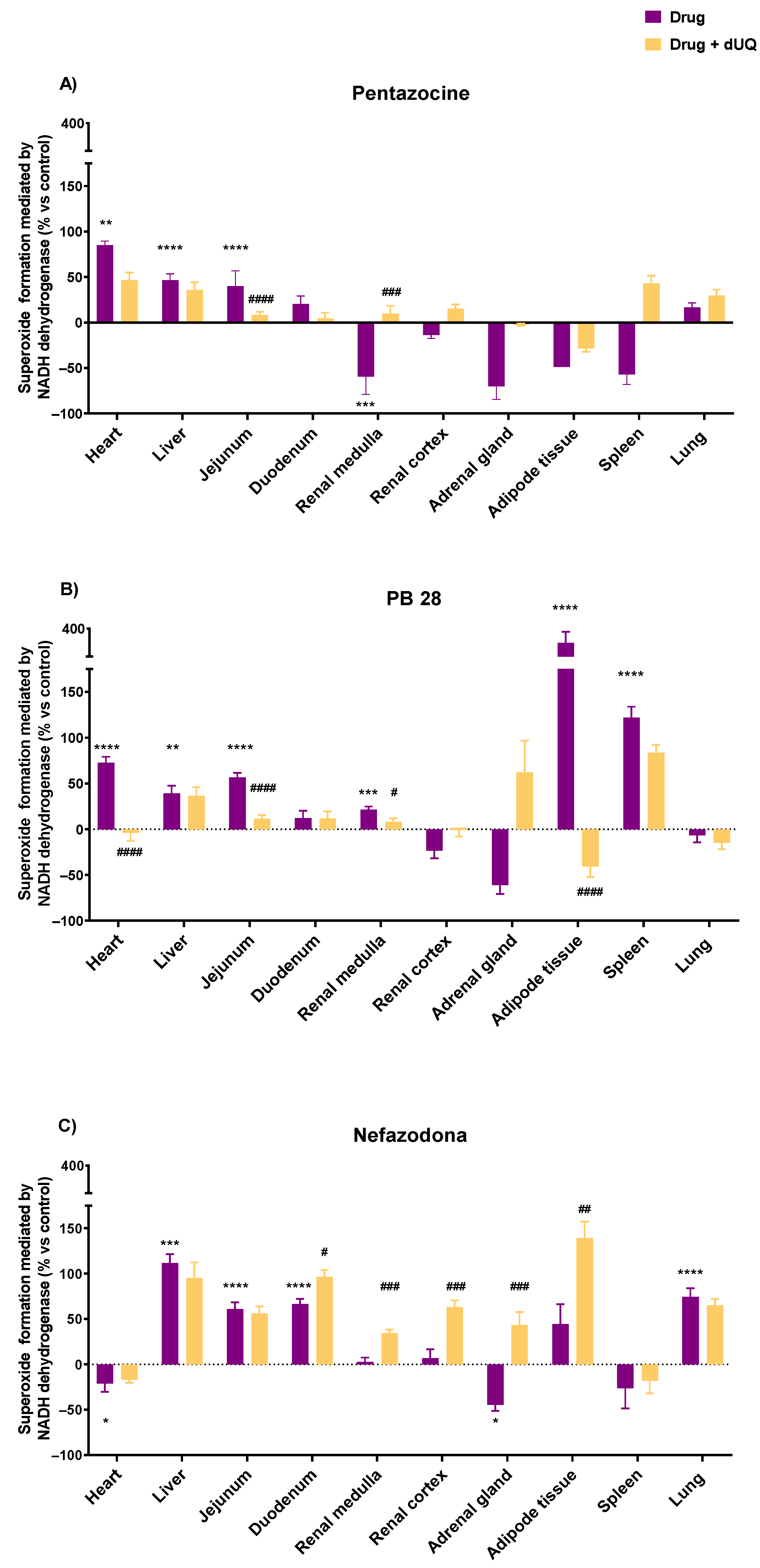
3.3. Tissue-Specific Effects of Drugs on Superoxide Formation Using CMMAs of Human Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Checa, J.; Aran, J.M. Reactive oxygen species: Drivers of physiological and pathological processes. J. Inflamm. Res. 2020, 13, 1057. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nature reviews. Immunology 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nature reviews. Neuroscience 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Do Carmo, L.S.; Berk, B.C.; Harrison, D.G. NOX5 as a therapeutic target in cerebral ischemic injury. J. Clin. Investig. 2019, 129, 1530–1532. [Google Scholar] [CrossRef]
- Tipton, K.F. 90 years of monoamine oxidase: Some progress and some confusion. J. Neural Transm. 2018, 125, 1519–1551. [Google Scholar] [CrossRef]
- Antonucci, S.; Di Sante, M.; Tonolo, F.; Pontarollo, L.; Scalcon, V.; Alanova, P.; Menabò, R.; Carpi, A.; Bindoli, A.; Rigobello, M.P.; et al. The determining role of mitochondrial reactive oxygen species generation and monoamine oxidase activity in doxorubicin-induced cardiotoxicity. Antioxid. Redox Signal. 2021, 34, 531–550. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, X.; Zhang, M.; Zhao, C.; Mei, C.; Li, P. MAPK pathway inhibitors attenuated hydrogen peroxide induced damage in neural cells. BioMed Res. Int. 2019, 2019, 5962014. [Google Scholar] [CrossRef]
- Angelova, P.R.; Dinkova-Kostova, A.T.; Abramov, A.Y. Assessment of ROS production in the mitochondria of live cells. Methods Mol. Biol. 2021, 2202, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shidara, Y.; Hollenbeck, P.J. Defects in mitochondrial axonal transport and membrane potential without increased reactive oxygen species production in a drosophila model of Friedreich ataxia. J. Neurosci. 2010, 30, 11369–11378. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Sigitova, E.; Fišar, Z.; Hroudová, J.; Cikánková, T.; Raboch, J. Biological hypotheses and biomarkers of bipolar disorder. Psychiatry Clin. Neurosci. 2017, 71, 77–103. [Google Scholar] [CrossRef]
- Kim, E.; Keskey, Z.; Kang, M.; Kitchen, C.; Bentley, W.E.; Chen, S.; Kelly, D.L.; Payne, G.F. Validation of oxidative stress assay for schizophrenia. Schizophr. Res. 2019, 212, 126–133. [Google Scholar] [CrossRef]
- Upthegrove, R.; Khandaker, G.M. Cytokines, oxidative stress and cellular markers of inflammation in schizophrenia. Curr. Top. Behav. Neurosci. 2020, 44, 49–66. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Rich, P.R.; Maréchal, A. The mitochondrial respiratory chain. Essays Biochem. 2010, 47, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, A.K.; Dhar, S.K.; Xu, Y.; Clair, D.K.S. Manganese superoxide dismutase: Beyond life and death. Amino Acids 2012, 42, 139–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hroudová, J.; Fišar, Z. In vitro inhibition of mitochondrial respiratory rate by antidepressants. Toxicol. Lett. 2012, 213, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.G.; Bandiera, S.M. Involvement of cytochrome P450 in reactive oxygen species formation and cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic. Biol. Med. 2007, 42, 926. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Chen, Y.; Clair, D.K.S. ROS and P53: Versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Clair, D.K.S. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Samal, R.R.; Kumari, K.; Sahoo, Y.; Mishra, S.K.; Subudhi, U. Interaction of artemisinin protects the activity of antioxidant enzyme catalase: A biophysical study. Int. J. Biol. Macromol. 2021, 172, 418–428. [Google Scholar] [CrossRef]
- Clay, H.B.; Sillivan, S.; Konradi, C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci. 2011, 29, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, W.T.; Buchanan, R.W. Schizophrenia. N. Engl. J. Med. 1994, 330, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current concepts and treatments of schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef]
- Cikánková, T.; Fišar, Z.; Bakhouche, Y.; Ľupták, M.; Hroudová, J. In vitro effects of antipsychotics on mitochondrial respiration. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 1209–1223. [Google Scholar] [CrossRef]
- Balijepalli, S.; Boyd, M.R.; Ravindranath, V. Inhibition of mitochondrial complex I by haloperidol: The role of thiol oxidation. Neuropharmacology 1999, 38, 567–577. [Google Scholar] [CrossRef]
- Park, H.J.; Choi, I.; Leem, K.H. Decreased brain PH and pathophysiology in schizophrenia. Int. J. Mol. Sci. 2021, 22, 8358. [Google Scholar] [CrossRef]
- Heiser, P.; Sommer, O.; Schmidt, A.J.; Clement, H.W.; Hoinkes, A.; Hopt, U.T.; Schulz, E.; Krieg, J.C.; Dobschütz, E. Effects of antipsychotics and vitamin C on the formation of reactive oxygen species. J. Psychopharmacol. 2010, 24, 1499–1504. [Google Scholar] [CrossRef]
- Heiss, K.; Raffaele, M.; Vanella, L.; Murabito, P.; Prezzavento, O.; Marrazzo, A.; Aricò, G.; Castracani, C.C.; Barbagallo, I.; Zappalà, A.; et al. (+)-Pentazocine attenuates SH-SY5Y cell death, oxidative stress and microglial migration induced by conditioned medium from activated microglia. Neurosci. Lett. 2017, 642, 86–90. [Google Scholar] [CrossRef]
- O’Brien, E.; Dedova, I.; Duffy, L.; Cordwell, S.; Karl, T.; Matsumoto, I. Effects of chronic risperidone treatment on the striatal protein profiles in rats. Brain Res. 2006, 1113, 24–32. [Google Scholar] [CrossRef]
- Muench, J.; Hamer, A.M. Adverse effects of antipsychotic medications. Am. Fam. Physician 2010, 81, 617–622. [Google Scholar] [PubMed]
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.I.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luca, M.; Di Mauro, M.; Di Mauro, M.; Luca, A. Gut microbiota in Alzheimer’s disease, depression, and type 2 diabetes mellitus: The role of oxidative stress. Oxidative Med. Cell. Longev. 2019, 2019, 4730539. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Barbosa, I.A.; Seabra, C.; Beltrão, N.; Santos, R.; Vega-Naredo, I.; Oliveira, P.J.; Cunha-Oliveira, T. Involvement of mitochondrial dysfunction in nefazodone-induced hepatotoxicity. Food Chem. Toxicol. 2016, 94, 148–158. [Google Scholar] [CrossRef]
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez Carranza, R. Clozapina: Antipsicóticos. In Vademécum Académico de Medicamentos; McGraw-Hill Education: New York, NY, USA, 2015. [Google Scholar]
- Stöllberger, C.; Huber, J.O.; Finsterer, J. Antipsychotic drugs and QT prolongation. Int. Clin. Psychopharmacol. 2005, 20, 243–251. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, R.; Ferreira, E.; Knowles, S.; Fux, C.; Rodin, A.; Winslow, W.; Oddo, S. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 2019, 18, e13037. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [Green Version]
- Pati, M.L.; Hornick, J.R.; Niso, M.; Berardi, F.; Spitzer, D.; Abate, C.; Hawkins, W. Sigma-2 receptor agonist derivatives of 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-Yl) propyl]piperazine (PB28) induce cell death via mitochondrial superoxide production and caspase activation in pancreatic cancer. BMC Cancer 2017, 17, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambunathan, N. Determination and detection of reactive oxygen species (ROS), lipid peroxidation, and electrolyte leakage in plants. Methods Mol. Biol. 2010, 639, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Elexpe, A.; Nieto, N.; Fernández-Cuétara, C.; Domínguez-Fernández, C.; Morera-Herreras, T.; Torrecilla, M.; Miguélez, C.; Laso, A.; Ochoa, E.; Bailen, M.; et al. Study of tissue-specific reactive oxygen species formation by cell membrane microarrays for the characterization of bioactive compounds. Membranes 2021, 11, 943. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Sánchez-Magraner, L.; De La Fuente, M.; Evans, C.; Miles, J.; Elexpe, A.; Rodriguez-Astigarraga, M.; Astigarraga, E.; Barreda-Gómez, G.; Samanidou, V.; Mandrioli, R.; et al. Quantification of PD-1/PD-L1 interaction between membranes from PBMCs and melanoma samples using cell membrane microarray and time-resolved Förster resonance energy transfer. Analytica 2021, 2, 156–170. [Google Scholar] [CrossRef]
- Rodriguez Puertas, R.; Barreda-Gómez, G.; Giralt Rue, M.T.; Fernández Pastor, B. Method of Quantifying the G Protein-Coupled Receptor (GPCR)/G Protein Coupling Using a Cell Membrane Array. U.S. Patent 11/996,965, 28 July 2006. [Google Scholar]
- Rienda, B.; Elexpe, A.; Tolentino-Cortez, T.; Gulak, M.; Bruzos-Cidón, C.; Torrecilla, M.; Astigarraga, E.; Barreda-Gómez, G. Analysis of acetylcholinesterase activity in cell membrane microarrays of brain areas as a screening tool to identify tissue specific inhibitors. Analytica 2021, 2, 25–36. [Google Scholar] [CrossRef]
- Manuel, I.; Barreda-Gomez, G.; González De San Román, E.; Veloso, A.; Fernández, J.A.; Giralt, M.T.; Rodriguez-Puertas, R. Neurotransmitter receptor localization: From autoradiography to imaging mass spectrometry. ACS Chem. Neurosci. 2015, 6, 362–373. [Google Scholar] [CrossRef]
- Fernández, R.; Garate, J.; Tolentino-Cortez, T.; Herraiz, A.; Lombardero, L.; Ducrocq, F.; Rodríguez-Puertas, R.; Trifilieff, P.; Astigarraga, E.; Barreda-Gómez, G.; et al. Microarray and mass spectrometry-based methodology for lipid profiling of tissues and cell cultures. Anal. Chem. 2019, 91, 15967–15973. [Google Scholar] [CrossRef]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Pagano Zottola, A.C.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef]
- Dietrich-Muszalska, A.; Malinowska, J.; Olas, B.; Głowacki, R.; Bald, E.; Wachowicz, B.; Rabe-Jabłońska, J. The oxidative stress may be induced by the elevated homocysteine in schizophrenic patients. Neurochem. Res. 2012, 37, 1057–1062. [Google Scholar] [CrossRef] [Green Version]
- Lepping, P.; Delieu, J.; Mellor, R.; Williams, J.H.H.; Hudson, P.R.; Hunter-Lavin, C. Antipsychotic medication and oxidative cell stress: A systematic review. J. Clin. Psychiatry 2011, 72, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Suárez, H.; Andreu, Z.; Mazzeo, C.; Toribio, V.; Pérez-Rivera, A.E.; López-Martín, S.; García-Silva, S.; Hurtado, B.; Morato, E.; Peláez, L.; et al. CD9 inhibition reveals a functional connection of extracellular vesicle secretion with mitophagy in melanoma cells. J. Extracell. Vesicles 2021, 10, e12082. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef]
- Salimi, A.; Razian, M.; Pourahmad, J. Analysis of toxicity effects of buspirone, cetirizine and olanzapine on human blood lymphocytes: In vitro model. Curr. Clin. Pharmacol. 2018, 13, 120–127. [Google Scholar] [CrossRef]
- Khorassani, F.; Saad, M. Intravenous olanzapine for the management of agitation: Review of the literature. Ann. Pharmacother. 2019, 53, 853–859. [Google Scholar] [CrossRef]
- Eftekhari, A.; Azarmi, Y.; Parvizpur, A.; Eghbal, M.A. Involvement of oxidative stress and mitochondrial/lysosomal cross-talk in olanzapine cytotoxicity in freshly isolated rat hepatocytes. Xenobiotica 2016, 46, 363–378. [Google Scholar] [CrossRef]
- Polydoro, M.; Schröder, N.; Lima, M.N.M.; Caldana, F.; Laranja, D.C.; Bromberg, E.; Roesler, R.; Quevedo, J.; Moreira, J.C.F.; Dal-Pizzol, F. Haloperidol- and clozapine-induced oxidative stress in the rat brain. Pharmacol. Biochem. Behav. 2004, 78, 751–756. [Google Scholar] [CrossRef]
- Reinke, A.; Martins, M.R.; Lima, M.S.; Moreira, J.C.; Dal-Pizzol, F.; Quevedo, J. Haloperidol and clozapine, but not olanzapine, induces oxidative stress in rat brain. Neurosci. Lett. 2004, 372, 157–160. [Google Scholar] [CrossRef]
- Martins, M.R.; Petronilho, F.C.; Gomes, K.M.; Dal-Pizzol, F.; Streck, E.L.; Quevedo, J. Antipsychotic-induced oxidative stress in rat brain. Neurotox. Res. 2008, 13, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Carranza, R. Pentazocina. Available online: https://www.vademecum.es/principios-activos-pentazocina-n02ad01 (accessed on 26 December 2021).
- Heiss, K.; Vanella, L.; Murabito, P.; Prezzavento, O.; Marrazzo, A.; Castruccio Castracani, C.; Barbagallo, I.; Zappalà, A.; Arena, E.; Astuto, M.; et al. (+)-Pentazocine reduces oxidative stress and apoptosis in microglia following hypoxia/reoxygenation injury. Neurosci. Lett. 2016, 626, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Natsvlishvili, N.; Goguadze, N.; Zhuravliova, E.; Mikeladze, D. Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria. BMC Biochem. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurogi, K.; Chen, M.; Lee, Y.; Shi, B.; Yan, T.; Liu, M.-Y.; Sakakibara, Y.; Suiko, M.; Liu, M.-C. Sulfation of buprenorphine, pentazocine, and naloxone by human cytosolic sulfotransferases. Drug Metab. Lett. 2012, 6, 109–115. [Google Scholar] [CrossRef]
- Appleyard, T.N. Effects of pentazocine on cardiac function. Proc. Roy. Soc. Med. 1975, 68, 770. [Google Scholar] [CrossRef] [Green Version]
- Lal, S.; Savidge, R.S.; Chhabra, G.P. Cardiovascular and respiratory effects of morphine and pentazocine in patients with myocardial infarction. Lancet 1969, 293, 379–381. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J.R. Drug-induced mitochondrial toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef]
- Balijepalli, S.; Kenchappa, R.S.; Boyd, M.R.; Ravindranath, V. Protein thiol oxidation by haloperidol results in inhibition of mitochondrial complex I in brain regions: Comparison with atypical antipsychotics. Neurochem. Int. 2001, 38, 425–435. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Neuroleptic medications inhibit complex I of the electron transport chain. Ann. Neurol. 1994, 35, 244–245. [Google Scholar] [CrossRef]
- Xu, F.; Xia, Y.; Feng, Z.; Lin, W.; Xue, Q.; Jiang, J.; Yu, X.; Peng, C.; Luo, M.; Yang, Y.; et al. Repositioning antipsychotic fluphenazine hydrochloride for treating triple negative breast cancer with brain metastases and lung metastases. Am. J. Cancer Res. 2019, 9, 459. [Google Scholar]
- Marsh, W. Nefazodone. Available online: https://pubmed.ncbi.nlm.nih.gov/31643507 (accessed on 17 March 2022).
- Sedky, K.; Nazir, R.; Joshi, A.; Kaur, G.; Lippmann, S. Which psychotropic medications induce hepatotoxicity? Gen. Hosp. Psychiatry 2012, 34, 53–61. [Google Scholar] [CrossRef] [PubMed]
- DeSanty, K.P.; Amabile, C.M. Antidepressant-induced liver injury. Ann. Pharmacother. 2007, 41, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.S.; Roberts, D.L.; Smith, J.M.; Stringfellow, J.C.; Kaplita, S.B.; Seminara, J.A.; Marcus, R.N. The safety profile of nefazodone. J. Clin. Psychiatry 1996, 57 (Suppl. 2), 31–38. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superoxide Formation | ||||
|---|---|---|---|---|
| Maximum Amount | Production Rate | |||
| Rotenone | pEC50 | −7.7 ± 0.23 | pIC50 | −7.47 ± 0.15 |
| Emax | 42.79 ± 4.88 | Vmax | 41.90 ± 2.94 | |
| Antimycin A | pEC50 | −5.94 ± 0.14 | pEC50 | −6.30 ± 0.35 |
| Emax | 34.20 ± 2.29 | Vmax | 21.04 ± 3.35 | |
| pEC50 | Emax (%) | pEC50 | Emax (%) | ||
|---|---|---|---|---|---|
| PB 28 | −5.4 ± 0.2 | 43.4 ± 3.0 | Pentzazocine | −3.9 ±0.3 | 45.5 ± 11.1 |
| NE 100 | −3.6 ± 0.3 | 60.1 ± 23.1 | Donepezil | UD | UD |
| PPCC | −4.5 ± 0.3 | 35.4 ± 5.3 | Fluphenazine | −4.6 ± 0.3 | 36.8 ± 4.9 |
| BD 1047 | −6.3 ± 0.4 | 20.6 ± 4.1 | Clozapine | UD | UD |
| DMT | UD | UD | Olanzapine | −5.5 ± 0.4 | 15.7 ± 3.1 |
| DXT | −3.9 ± 0.3 | 19.0 ± 5.2 | Desclozapine | −3.2 ± 1.0 | UD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elexpe, A.; Sánchez-Sánchez, L.; Tolentino-Cortez, T.; Astigarraga, E.; Torrecilla, M.; Barreda-Gómez, G. Analysis of Mitochondrial Function in Cell Membranes as Indicator of Tissue Vulnerability to Drugs in Humans. Biomedicines 2022, 10, 980. https://doi.org/10.3390/biomedicines10050980
Elexpe A, Sánchez-Sánchez L, Tolentino-Cortez T, Astigarraga E, Torrecilla M, Barreda-Gómez G. Analysis of Mitochondrial Function in Cell Membranes as Indicator of Tissue Vulnerability to Drugs in Humans. Biomedicines. 2022; 10(5):980. https://doi.org/10.3390/biomedicines10050980
Chicago/Turabian StyleElexpe, Ane, Laura Sánchez-Sánchez, Tarson Tolentino-Cortez, Egoitz Astigarraga, María Torrecilla, and Gabriel Barreda-Gómez. 2022. "Analysis of Mitochondrial Function in Cell Membranes as Indicator of Tissue Vulnerability to Drugs in Humans" Biomedicines 10, no. 5: 980. https://doi.org/10.3390/biomedicines10050980