
Vasoconstrictor and Pressor Effects of Des-Aspartate-Angiotensin I in Rat

 ,
,  , ,
, ,  ,
,  , and
, and 
Abstract
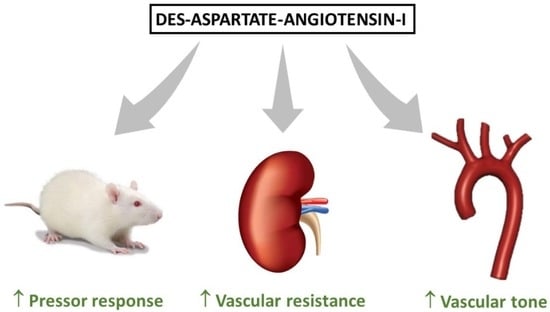
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Protocols
2.2.1. Experiment 1. Whole Body Response to DAA-I and Ang II
2.2.2. Experiment 2. Renal Response to DAA-I and Ang II in the Isolated Rat Kidney
2.2.3. Experiment 3. Response to DAA-I and Ang II in the Presence and Absence of Endothelium in Aortic Rings
2.3. Drugs
2.4. Statistical Analyses
3. Results
3.1. Whole Body Response
3.2. Vasoconstrictor Response in the Renal Vasculature
3.3. Effect on Aortic Ring Tone
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar] [PubMed]
- Paul, M.; Mehr, A.R.; Kreutz, R. Physiology of local renin–angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Chappell, M.C.; Tallant, E.A.; Brosnihan, K.B.; Diz, D.I. Counterregulatory actions of angiotensin-(1–7). Hypertension 1997, 30, 535–541. [Google Scholar] [CrossRef]
- Skeggs, L.T.; Lentz, K.E.; Gould, A.B.; Hochstrasser, H.; Kahn, J.R. Biochemistry and kinetics of the renin–angiotensin system. Fed. Proc. 1967, 26, 42–47. [Google Scholar] [PubMed]
- Jankowski, V.; Vanholder, R.; van der Giet, M.; Tölle, M.; Karadogan, S.; Gobom, J.; Furkert, J.; Oksche, A.; Krause, E.; Tran, T.N.; et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Chansel, D.; Ardaillou, R. Active metabolites derived from angiotensin II. Nephrologie 1998, 19, 427–432. [Google Scholar]
- Mustafa, T.; Lee, J.H.; Chai, S.Y.; Albiston, A.L.; McDowall, S.G.; Mendelsohn, F.A. Bioactive angiotensin peptides: Focus on angiotensin IV. J. Renin Angiotensin Aldosterone Syst. 2001, 2, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Smolders, I.; Dupont, A.G. Blood pressure and renal hemodynamic effects of angiotensin fragments. Hypertens. Res. 2011, 34, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, C.G.; Smellie, W.S.; Morton, J.J. Des-Asp-angiotensin I: Its identification in rat blood and confirmation as a substrate for converting enzyme. Endocrinology 1981, 108, 406–412. [Google Scholar] [CrossRef]
- Campbell, D.J.; Lawrence, A.C.; Towrie, A.; Kladis, A.; Valentijn, A.J. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension 1991, 18, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Sim, M.K.; Qui, X.S. Angiotensins in plasma of hypertensive rats and human. Regul. Pept. 2003, 111, 179–182. [Google Scholar] [CrossRef]
- Sim, M.K. Des-aspartate-angiotensin I, a novel angiotensin AT(1) receptor drug. Eur. J. Pharmacol. 2015, 760, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sethi, G.; Loke, W.K.; Sim, M.K. Des-Aspartate-Angiotensin I Attenuates Mortality of Mice Exposed to Gamma Radiation via a Novel Mechanism of Action. PLoS ONE 2015, 10, e0138009. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.O.; Khoo, C.M.; Chowbay, B.; Chan, Y.H.; Sim, M.K. A Single Dose-Escalation Study to Evaluate the Safety and Pharmacokinetics of Orally Administered Des-Aspartate Angiotensin I in Healthy Subjects. Drugs R D 2016, 16, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.O.; Tian, E.F.; Cai, M.H.; Wang, H.; Chan, Y.H.; Sim, M.K. Bioavailability of Orally Administered Des-Aspartate-Angiotensin I in Human Subjects. Drugs R D 2018, 18, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Kono, T.; Ikeda, F.; Oseko, F.; Imura, H.; Endo, J. Biological activity of des-Asp1-angiotensin I in man. J. Clin. Endocrinol. Metab. 1980, 50, 40–45. [Google Scholar] [CrossRef]
- Lim, B.C.; Sim, M.K. Actions of des-Asp angiotensin I on the aortic rings of the normo- and hypertensive rats. Clin. Exp. Hypertens. 1998, 20, 105–117. [Google Scholar] [CrossRef]
- Sim, M.K.; Yuan, H.T. Effect of des-Asp-angiotensin I on the contractile action of angiotensin II and angiotensin III. Eur. J. Pharmacol. 1995, 258, 175–178. [Google Scholar] [CrossRef]
- Mustafa, M.R.; Dharmani, M.; Kunheen, N.K.; Sim, M.K. Effects of des-aspartate-angiotensin I on the actions of angiotensin III in the renal and mesenteric vasculature of normo- and hypertensive rats. Regul. Pept. 2004, 120, 15–22. [Google Scholar] [CrossRef]
- Sim, M.K.; Radhakrishnan, R. Novel central action of des-Asp-angiotensin I. Eur. J. Pharmacol. 1994, 257, R1–R3. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Sim, M.K. Actions of D-amino acid-substituted analogues of des-Asp-angiotensin I on the central pressor action of angiotensin III. Eur. J. Pharmacol. 1995, 294, 337–339. [Google Scholar] [CrossRef]
- Dharmani, M.; Mustafa, M.R.; Achike, F.I.; Sim, M.K. Effect of des-aspartate-angiotensin I on the actions of angiotensin II in the isolated renal and mesenteric vasculature of hypertensive and STZ-induced diabetic rats. Regul. Pept. 2005, 129, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Vargas, F.; Osuna, A. Modulatory role of endothelium-derived relaxing factors on the response to vasoconstrictors and flow–pressure curve in the isolated perfused rat kidney. J. Vasc. Res. 1996, 33, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, R.; López-Sepúlveda, R.; Kadmiri, M.; Romero, M.; Vera, R.; Sánchez, M.; Vargas, F.; O’Valle, F.; Zarzuelo, A.; Dueñas, M.; et al. Polyphenols restore endothelial function in DOCA-salt hypertension: Role of endothelin-1 and NADPH oxidase. Free Radic. Biol. Med. 2007, 43, 462–473. [Google Scholar] [CrossRef]
- Cowley, A.W. Long-term control of arterial pressure. Physiol Rev. 1992, 272, 211–300. [Google Scholar] [CrossRef]
- Moreno, J.M.; Gómez, I.R.; Wangensteen, R.; Guerra, M.A.; de Dios Luna, J.; García-Estañ, J.; Vargas, F. Tempol improves renal hemodynamics and pressure natriuresis in hyperthyroid rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R867–R873. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.Y.; DeWitt, B.J.; McMahon, T.J.; Kadowitz, P.J. Comparison of pressor responses to angiotensin I, II, and III in pulmonary vascular bed of cats. Am. J. Physiol. 1994, 266, H2247–H2255. [Google Scholar] [CrossRef]
- Regoli, D.; Park, W.K.; Rioux, F. Pharmacology of angiotensin. Pharmacol. Rev. 1974, 26, 69–123. [Google Scholar]
- Peach, M.J. Renin-angiotensin system: Biochemistry and mechanisms of action. Physiol. Rev. 1977, 57, 313–370. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart. Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Hussain, M.; Awan, F.R. Hypertension regulating angiotensin peptides in the pathobiology of cardiovascular disease. Clin. Exp. Hypertens. 2018, 40, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zhang, J.; Zhuo, J.L. The vasoprotective axes of the renin-angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacol. Res. 2017, 125, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Sim, M.K.; Xu, X.G. Effects of des-aspartate-angiotensin I on angiotensin II-induced incorporation of phenylalanine and thymidine in cultured rat cardiomyocytes and aortic smooth muscle cells. Regul. Pept. 2000, 95, 93–97. [Google Scholar] [CrossRef]
- Loh, W.M.; Ling, W.C.; Murugan, D.D.; Lau, Y.S.; Achike, F.I.; Vanhoutte, P.M.; Mustafa, M.R. Des-aspartate angiotensin I (DAA-I) reduces endothelial dysfunction in the aorta of the spontaneously hypertensive rat through inhibition of angiotensin II-induced oxidative stress. Vascul. Pharmacol. 2015, 71, 151–158. [Google Scholar] [CrossRef]
- Wen, Q.; Lee, K.O.; Sim, S.Z.; Xu, X.G.; Sim, M.K. Des-aspartate-angiotensin I causes specific release of PGE2 and PGI2 in HUVEC via the angiotensin AT1 receptor and biased agonism. Eur. J. Pharmacol. 2015, 768, 173–181. [Google Scholar] [CrossRef]
- Dharmani, M.; Mustafa, M.R.; Achike, F.I.; Sim, M.K. Involvement of AT(1) angiotensin receptors in the vasomodulatory effect of des-aspartate-angiotensin I in the rat renal vasculature. Peptides 2008, 29, 1773–1780. [Google Scholar] [CrossRef] [Green Version]
- Sim, M.K.; Soh, K.S. Effects of des-Asp-angiotensin I on the electrically stimulated contraction of the rabbit pulmonary artery. Eur. J. Pharmacol. 1995, 284, 215–219. [Google Scholar] [CrossRef]
- Gammelgaard, I.; Wamberg, S.; Bie, P. Systemic effects of angiotensin III in conscious dogs during acute double blockade of the renin–angiotensin–aldosterone-system. Acta Physiol. 2006, 188, 129–138. [Google Scholar] [CrossRef]
- Pendleton, R.G.; Gessner, G.; Horner, E. Comparative effects of angiotensin II and angiotensin III in rabbit adrenal and aortic tissues. J. Pharmacol. Exp. Ther. 1991, 256, 614–620. [Google Scholar]
- Healy, D.P.; Song, L. Kidney aminopeptidase A and hypertension, part I: Spontaneously hypertensive rats. Hypertension 1999, 33, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, E.D.; Peach, M.J.; Ackerly, J.A.; Tsai, B.S.; Larner, A. Pressor and stereoidogenic actions of [des-Asp1]angiotensin I dependency on conversion to angiotensin III. Circ. Res. 1977, 40, I94–I97. [Google Scholar] [PubMed]
- Velez, J.C.; Janech, M.G.; Hicks, M.P.; Morinelli, T.A.; Rodgers, J.; Self, S.E.; Arthur, J.M.; Fitzgibbon, W.R. Lack of renoprotective effect of chronic intravenous angiotensin-(1-7) or angiotensin-(2-10) in a rat model of focal segmental glomerulosclerosis. PLoS ONE 2014, 9, e110083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, R.; Friedrich, C.; Wolbers, M.; Mann, J.F. Meta-analysis: Effect of monotherapy and combination therapy with inhibitors of the renin–angiotensin system on proteinuria in renal disease. Ann. Intern. Med. 2008, 148, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Huang, R.; Kavanagh, J.; Li, L.; Zeng, X.; Li, Y.; Fu, P. Efficacy and safety of dual blockade of the renin–angiotensin–aldosterone system in diabetic kidney disease: A meta-analysis. Am. J. Cardiovasc. Drugs 2019, 19, 259–286. [Google Scholar] [CrossRef]
- Yang, H.Y.; Erdös, E.G.; Levin, Y. A dipeptidyl carboxypeptidase that converts angiotensin I and inactivates bradykinin. Biochem. Biophys. Acta 1970, 214, 374–376. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wangensteen, R.; Gómez-Guzmán, M.; Banegas, I.; Rodríguez-Gómez, I.; Jiménez, R.; Duarte, J.; García-Estañ, J.; Vargas, F. Vasoconstrictor and Pressor Effects of Des-Aspartate-Angiotensin I in Rat. Biomedicines 2022, 10, 1230. https://doi.org/10.3390/biomedicines10061230
Wangensteen R, Gómez-Guzmán M, Banegas I, Rodríguez-Gómez I, Jiménez R, Duarte J, García-Estañ J, Vargas F. Vasoconstrictor and Pressor Effects of Des-Aspartate-Angiotensin I in Rat. Biomedicines. 2022; 10(6):1230. https://doi.org/10.3390/biomedicines10061230
Chicago/Turabian StyleWangensteen, Rosemary, Manuel Gómez-Guzmán, Inmaculada Banegas, Isabel Rodríguez-Gómez, Rosario Jiménez, Juan Duarte, Joaquín García-Estañ, and Félix Vargas. 2022. "Vasoconstrictor and Pressor Effects of Des-Aspartate-Angiotensin I in Rat" Biomedicines 10, no. 6: 1230. https://doi.org/10.3390/biomedicines10061230