
The Potential Application and Promising Role of Targeted Therapy in Pulmonary Arterial Hypertension

 , , , , and
, , , , and
Abstract
:1. Background
2. The Etiology of PAH
2.1. Physiopathogenesis of PAH
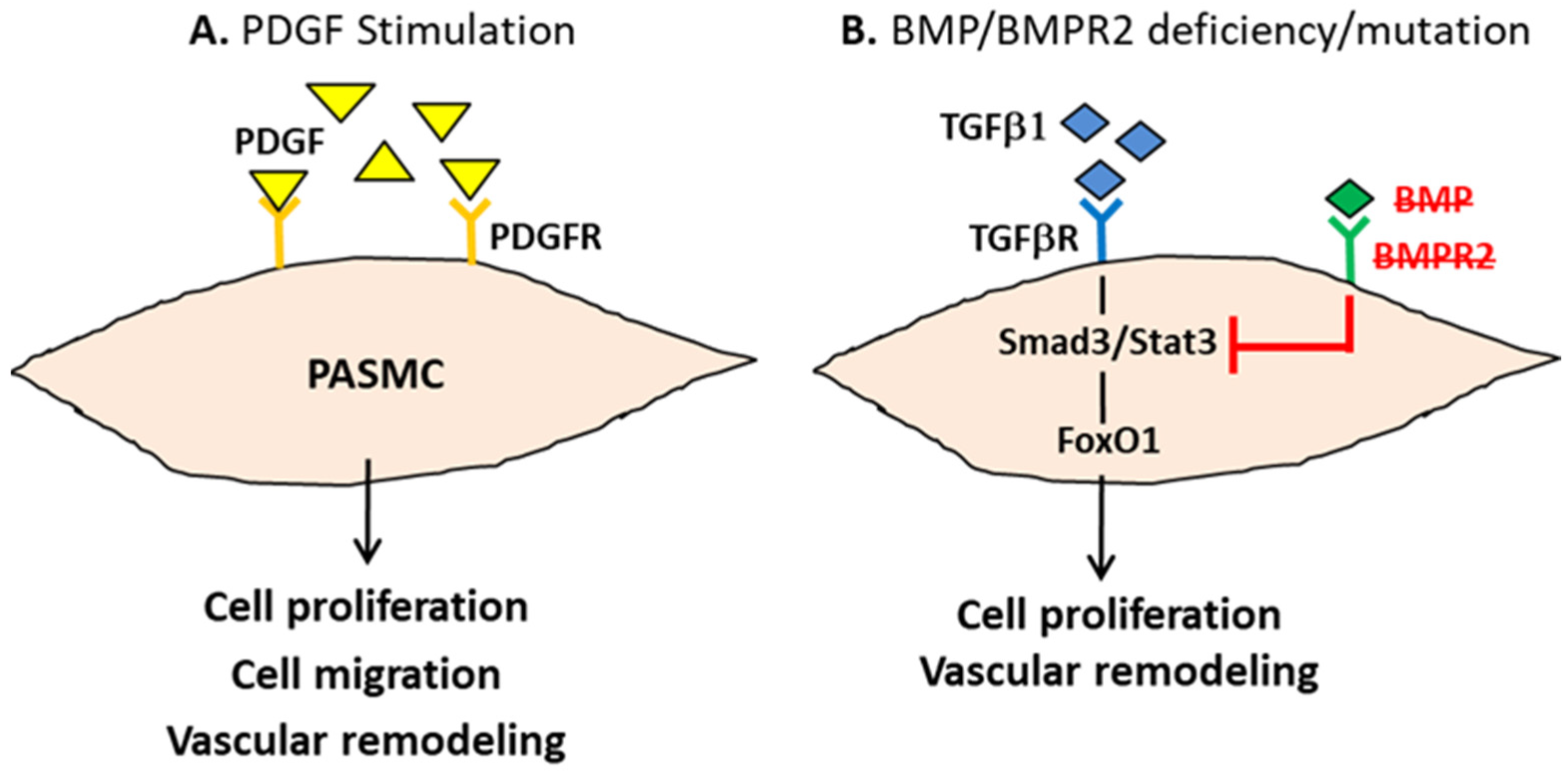
2.2. The Molecular Basis of PAH Pathogenesis
3. Similarities between PAH and Cancer
3.1. Warburg Effect
3.2. Mitochondrial Dysfunction
4. Current Strategies in PAH Therapeutics
4.1. Conventional Treatment
4.2. Background Therapies
5. Targeted Therapies in PAH Therapeutics
5.1. Introduction
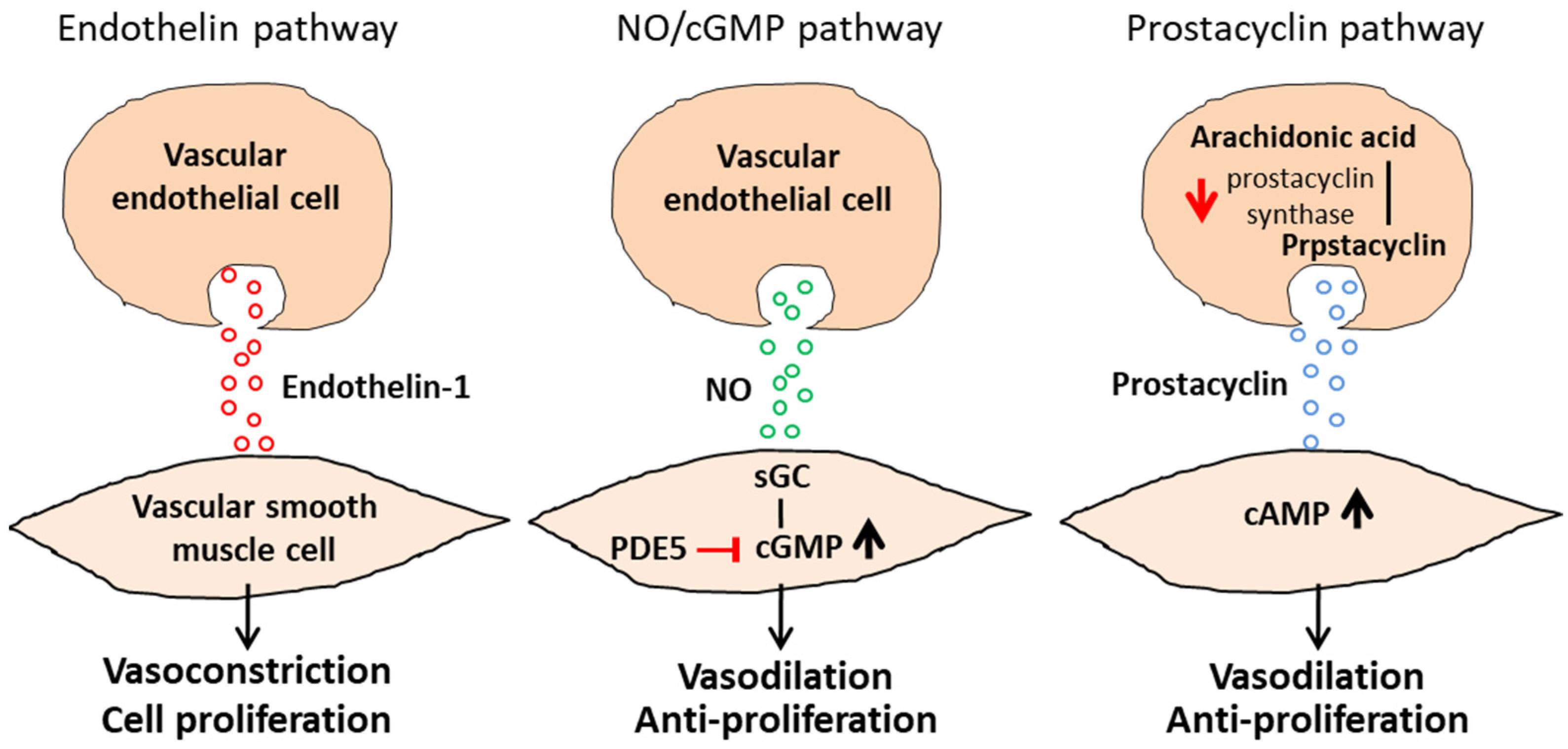
5.2. Pharmacological Agents for the Three Major Cellular Signaling Pathways
5.3. Calcium-Channel Blockade
5.4. The Inadequacy of Current PAH Management
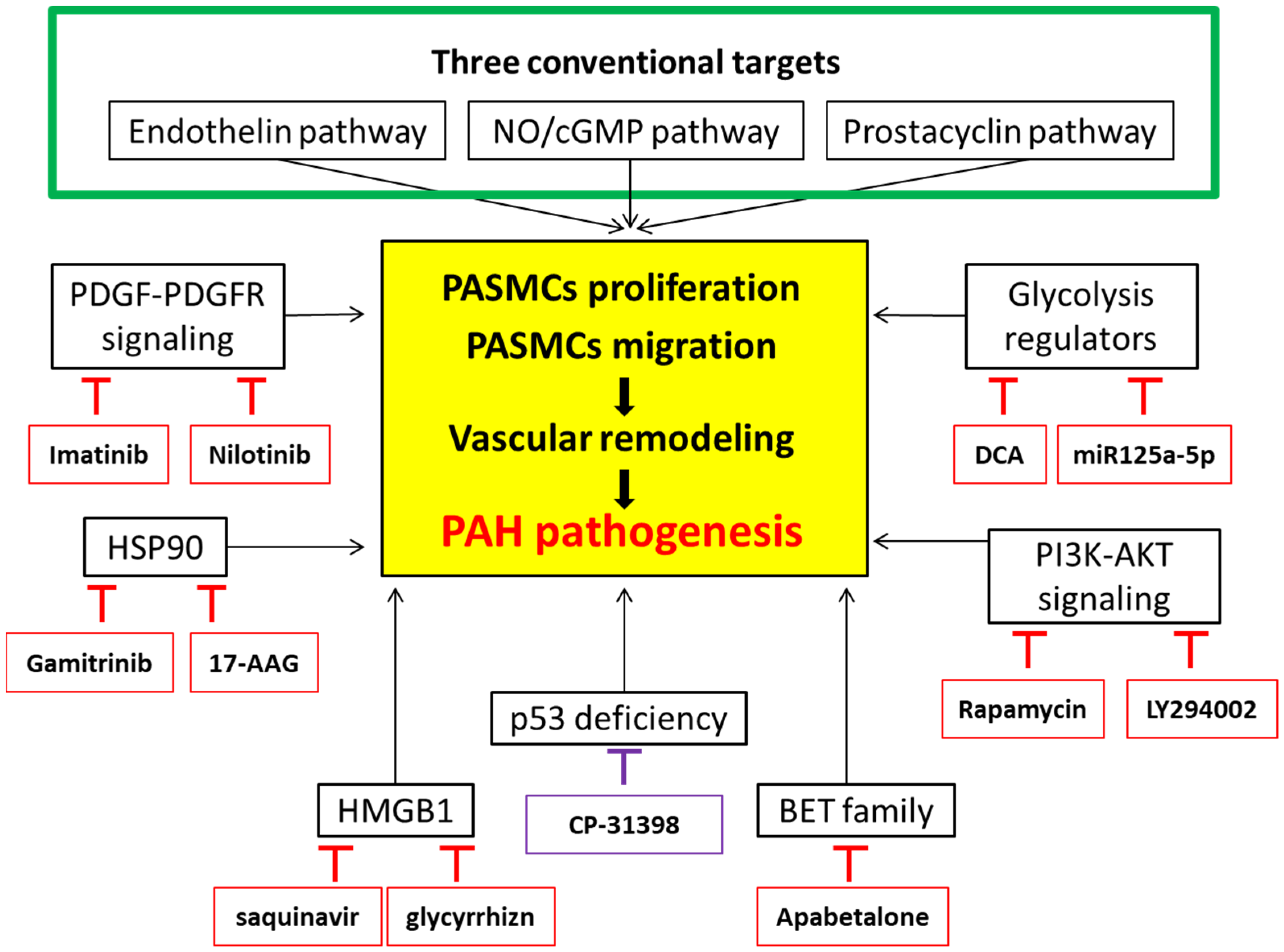
6. Targets under Consideration
6.1. Platelet-Derived Growth Factor Receptor
6.2. PI3K/AKT Signaling Cascades
6.3. Regulators in Glycolytic Metabolism
6.4. Heat Shock Protein (HSP) 90
6.5. High-Mobility Group Box-1 (HMGB1)
6.6. Bromodomain and Extra-Terminal (BET) Proteins
6.7. p53
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Morrell, N.; Archer, S.L.; Stenmark, K.R.; MacLean, M.; Lang, I.M.; Christman, B.W.; Weir, E.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, A.E.; Saggar, R.; Channick, R.N. Update on Medical Management of Pulmonary Arterial Hypertension. Cardiol. Clin. 2021, 40, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; McLaughlin, V.V.; Rubin, L.J.; Simonneau, G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1802148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Li, L.; Jick, S.; Breitenstein, S.; Hernandez, G.; Michel, A.; Vizcaya, D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm. Circ. 2017, 7, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Benza, R.L.; Miller, D.P.; Gomberg-Maitland, M.; Frantz, R.P.; Foreman, A.J.; Coffey, C.S.; Frost, A.E.; Barst, R.J.; Badesch, D.B.; Elliott, C.G.; et al. Predicting survival in pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation 2010, 122, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Kurakula, K.; Smolders, V.F.E.D.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial dysfunction in pulmonary hypertension: Cause or consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef]
- Santos-Ribeiro, D.; Mendes-Ferreira, P.; Maia-Rocha, C.; Adão, R.; Leite-Moreira, A.F.; Brás-Silva, C. Pulmonary arterial hypertension: Basic knowledge for clinicians. Arch. Cardiovasc. Dis. 2016, 109, 550–561. [Google Scholar] [CrossRef]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. J. Cereb. Blood Flow Metab. 2020, 178, 6–30. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, Z.G.S.; Klinger, J.R. Guidelines for the Treatment of Pulmonary Arterial Hypertension. Lung 2020, 198, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S. Sildenafil for pulmonary arterial hypertension: Exciting, but protection required. Chest 2003, 123, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Wharton, J.; Strange, J.W.; Møller, G.M.O.; Growcott, E.J.; Ren, X.; Franklyn, A.P.; Phillips, S.C.; Wilkins, M.R. Antiproliferative Effects of Phosphodiesterase Type 5 Inhibition in Human Pulmonary Artery Cells. Am. J. Respir. Crit. Care Med. 2005, 172, 105–113. [Google Scholar] [CrossRef]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An Imbalance between the Excretion of Thromboxane and Prostacyclin Metabolites in Pulmonary Hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef]
- Zhao, F.-Y.; Xu, S.-L.; Zhang, C.-F.; Liu, J.; Zhang, Y.; Yang, J.; Xing, X.-Q. PDGF mediates pulmonary arterial smooth muscle cell proliferation and migration by regulating NFATc2. Mol. Med. Rep. 2020, 23, 39. [Google Scholar] [CrossRef]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ links BMP2 and TGFβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef] [Green Version]
- Thenappan, T.; Ormiston, M.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Paulin, R.; Michelakis, E.D. The Metabolic Theory of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate Prevents and Reverses Pulmonary Hypertension by Inducing Pulmonary Artery Smooth Muscle Cell Apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.J.; Marsboom, G.; Fang, Y.-H.; Toth, P.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. PGC1α-mediated Mitofusin-2 Deficiency in Female Rats and Humans with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung18F-Fluorodeoxyglucose Positron Emission Tomography for Diagnosis and Monitoring of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef] [Green Version]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.-I.; et al. BMPR2 Preserves Mitochondrial Function and DNA during Reoxygenation to Promote Endothelial Cell Survival and Reverse Pulmonary Hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Garg, S.; Khunger, M.; Garg, S.; Kumbhani, D.J.; Chin, K.M.; Berry, J.D. Efficacy and safety of exercise training in chronic pulmonary hypertension: Systematic review and meta-analysis. Circ. Heart Fail. 2015, 8, 1032–1043. [Google Scholar] [CrossRef]
- Ulrich, S.; Hasler, E.D.; Saxer, S.; Furian, M.; Müller-Mottet, S.; Keusch, S.; Bloch, K.E. Effect of breathing oxygen-enriched air on exercise performance in patients with precapillary pulmonary hypertension: Randomized, sham-controlled cross-over trial. Eur. Heart J. 2017, 38, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Jais, X.; Olsson, K.M.; Barberà, J.A.; Blanco, I.; Torbicki, A.; Peacock, A.; Vizza, C.D.; Macdonald, P.; Humbert, M.; Hoeper, M.M. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur. Respir. J. 2012, 40, 881–885. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Kiely, D.G.; Cockrill, B.A.; Safdar, Z.; Wilson, V.J.; Al Hazmi, M.; Preston, I.R.; MacLean, M.R.; Lahm, T. Statement on Pregnancy in Pulmonary Hypertension from the Pulmonary Vascular Research Institute. Pulm. Circ. 2015, 5, 435–465. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, S.; Saxer, S.; Hasler, E.D.; Schwarz, E.I.; Schneider, S.R.; Furian, M.; Bader, P.R.; Lichtblau, M.; Bloch, K.E. Effect of domiciliary oxygen therapy on exercise capacity and quality of life in patients with pulmonary arterial or chronic thromboembolic pulmonary hypertension: A randomised, placebo-controlled trial. Eur. Respir. J. 2019, 54, 1900276. [Google Scholar] [CrossRef] [PubMed]
- Rich, S.; Seidlitz, M.; Dodin, E.; Osimani, D.; Judd, D.; Genthner, D.; McLaughlin, V.; Francis, G. The Short-term Effects of Digoxin in Patients with Right Ventricular Dysfunction From Pulmonary Hypertension. Chest 1998, 114, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V.; Steele, P.M.; Edwards, W.D.; Gersh, B.J.; McGoon, M.D.; Frye, R.L. Primary pulmonary hypertension: Natural history and the importance of thrombosis. Circulation 1984, 70, 580–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, H.; Ruber, K.; Mlczoch, J.; Schuster, E.; Gurtner, H.P.; Kneussl, M. The Effect of Anticoagulant Therapy in Primary and Anorectic Drug-Induced Pulmonary Hypertension. Chest 1997, 112, 714–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.M.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar]
- Preston, I.R.; Roberts, K.E.; Miller, D.P.; Sen, G.P.; Selej, M.; Benton, W.W.; Hill, N.S.; Farber, H.W. Effect of Warfarin Treatment on Survival of Patients with Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015, 132, 2403–2411. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Tsai, K.-B.; Hsu, J.-H.; Shin, S.-J.; Wu, J.-R.; Yeh, J.-L. Liraglutide prevents and reverses monocrotaline-induced pulmonary arterial hypertension by suppressing ET-1 and enhancing eNOS/sGC/PKG pathways. Sci. Rep. 2016, 6, 31788. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.C.; Wang, W.T.; Lee, S.S.; Kuo, Y.R.; Wang, Y.C.; Yen, S.J.; Lee, M.Y.; Yeh, J.L. Glucagon-like peptide-1 receptor agonist attenuates autophagy to ameliorate pulmonary arterial hypertension through Drp1/NOS- and Atg-5/Atg-7/Beclin-1/LC3β pathways. Int. J. Mol. Sci. 2019, 20, 3435. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Humbert, M.; Jais, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-Term Response to Calcium Channel Blockers in Idiopathic Pulmonary Arterial Hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- Rich, S.; Kaufmann, E.; Levy, P.S. The Effect of High Doses of Calcium-Channel Blockers on Survival in Primary Pulmonary Hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef]
- Montani, D.; Savale, L.; Natali, D.; Jaïs, X.; Herve, P.; Garcia, G.; Humbert, M.; Simonneau, G.; Sitbon, O. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur. Heart J. 2010, 31, 1898–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeper, M.M.; Apitz, C.; Grünig, E.; Halank, M.; Ewert, R.; Kaemmerer, H.; Kabitz, H.-J.; Kähler, C.; Klose, H.; Leuchte, H.; et al. Targeted therapy of pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Ghofrani, H.A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016, 71, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Norman, P. Evaluation of WO-2014132220, selective PDGFR inhibitors for the treatment of pulmonary arterial hypertension. Expert Opin. Ther. Patents 2015, 25, 493–499. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, J.; Qi, X.; Tao, X.; Xie, W.; Wan, J.; Shen, Y.H.; Zhai, Z. Plasminogen activator Inhibitor-2 inhibits pulmonary arterial smooth muscle cell proliferation in pulmonary arterial hypertension via PI3K/Akt and ERK signaling. Exp. Cell Res. 2020, 398, 112392. [Google Scholar] [CrossRef]
- Hu, Z.; Song, Q.; Ma, H.; Guo, Y.; Zhang, T.; Xie, H.; Luo, X. TRIM32 inhibits the proliferation and migration of pulmonary artery smooth muscle cells through the inactivation of PI3K/Akt pathway in pulmonary arterial hypertension. J. Bioenerg. Biomembr. 2021, 53, 309–320. [Google Scholar] [CrossRef]
- Leong, Z.P.; Hikasa, Y. Effects of toceranib compared with sorafenib on monocrotaline-induced pulmonary arterial hypertension and cardiopulmonary remodeling in rats. Vasc. Pharmacol. 2018, 110, 31–41. [Google Scholar] [CrossRef]
- Tang, H.; Chen, J.; Fraidenburg, D.R.; Song, S.; Sysol, J.R.; Drennan, A.R.; Offermanns, S.; Ye, R.D.; Bonini, M.G.; Minshall, R.D.; et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L208–L220. [Google Scholar] [CrossRef] [Green Version]
- Babicheva, A.; Makino, A.; Yuan, J. mTOR Signaling in Pulmonary Vascular Disease: Pathogenic Role and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 2144. [Google Scholar] [CrossRef]
- Houssaini, A.; Abid, S.; Mouraret, N.; Wan, F.; Rideau, D.; Saker, M.; Marcos, E.; Tissot, C.-M.; Dubois-Randé, J.-L.; Amsellem, V.; et al. Rapamycin Reverses Pulmonary Artery Smooth Muscle Cell Proliferation in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Peng, H.; Hong, C.; Chen, Z.; Deng, X.; Wang, A.; Yang, F.; Yang, L.; Chen, C.; Qin, X. PDGF Promotes the Warburg Effect in Pulmonary Arterial Smooth Muscle Cells via Activation of the PI3K/AKT/mTOR/HIF-1α Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Suda, M.; Katsuumi, G.; Nakao, M.; Hoyano, M.; Kashimura, T.; et al. Role of smooth muscle cell p53 in pulmonary arterial hypertension. PLoS ONE 2019, 14, e0212889. [Google Scholar] [CrossRef]
- Luo, L.; Xiao, L.; Lian, G.; Wang, H.; Xie, L. miR-125a-5p inhibits glycolysis by targeting hexokinase-II to improve pulmonary arterial hypertension. Aging 2020, 12, 9014–9030. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhao, R.; Liu, Q.; Li, Q. New Insights Into Heat Shock Protein 90 in the Pathogenesis of Pulmonary Arterial Hypertension. Front. Physiol. 2020, 11, 1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, S.; Zhao, Z.-M.; Liu, S.-X.; Zhang, G.-X.; Yang, F.; Wang, Y.; Wu, F.; Zhao, X.-X.; Xu, Z.-Y. Inhibition of heat shock protein 90 improves pulmonary arteriole remodeling in pulmonary arterial hypertension. Oncotarget 2016, 7, 54263–54273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 Accumulation Promotes Vascular Remodeling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Wang, J.; Yan, X.; Zhang, Q.; Chai, L.; Wang, Q.; Shi, W.; Chen, Y.; Liu, J.; Qu, Z.; et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. 2021, 54, e13048. [Google Scholar] [CrossRef]
- Wang, J.; Tian, X.-T.; Peng, Z.; Li, W.-Q.; Cao, Y.-Y.; Li, Y.; Li, X.-H. HMGB1/TLR4 promotes hypoxic pulmonary hypertension via suppressing BMPR2 signaling. Vasc. Pharmacol. 2019, 117, 35–44. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Futur. Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Potus, F.; Vaillancourt, M.; Bourgeois, A.; Johnson, I.; Deschamps, L.; Chabot, S.; Ruffenach, G.; Henry, S.; Breuils-Bonnet, S.; et al. Bromodomain-containing protein 4: The epigenetic origin of pulmonary arterial hypertension. Circ. Res. 2015, 117, 525–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L.; Szulcek, R.; Bourgeois, A.; Lampron, M.-C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Hennigs, J.K.; Cao, A.; Li, C.G.; Shi, M.; Mienert, J.; Miyagawa, K.; Körbelin, J.; Marciano, D.P.; Chen, P.-I.; Roughley, M.; et al. PPARγ-p53-Mediated Vasculoregenerative Program to Reverse Pulmonary Hypertension. Circ. Res. 2021, 128, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Bogaard, H.J.; Kraskauskas, D.; Alhussaini, A.; Gomez-Arroyo, J.; Voelkel, N.F.; Ishizaki, T. p53 Gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L753–L761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquin, S.; Rincheval, V.; Mignotte, B.; Richard, S.; Humbert, M.; Mercier, O.; Londoño-Vallejo, A.; Fadel, E.; Eddahibi, S. Inactivation of p53 Is Sufficient to Induce Development of Pulmonary Hypertension in Rats. PLoS ONE 2015, 10, e0131940. [Google Scholar] [CrossRef]
- Kochhar, A.; Kopelovich, L.; Sue, E.; Guttenplan, J.B.; Herbert, B.-S.; Dannenberg, A.J.; Subbaramaiah, K. p53 Modulates Hsp90 ATPase Activity and Regulates Aryl Hydrocarbon Receptor Signaling. Cancer Prev. Res. 2014, 7, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Zehendner, C.M.; Valasarajan, C.; Werner, A.; Boeckel, J.-N.; Bischoff, F.C.; John, D.; Weirick, T.; Glaser, S.F.; Rossbach, O.; Jaé, N.; et al. Long Noncoding RNA TYKRIL Plays a Role in Pulmonary Hypertension via the p53-mediated Regulation of PDGFRβ. Am. J. Respir. Crit. Care Med. 2020, 202, 1445–1457. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| PASMCs in PAH | Cancer Cells | |
|---|---|---|
| Cell proliferation | ↑ | ↑ |
| Cell migration | ↑ | ↑ |
| Cell apoptosis | ↓ | ↓ |
| Glycolysis | ↑ | ↑ |
| Fatty acid synthesis | ↑ | ↑ |
| Glutamine metabolism | ↑ | ↑ |
| Mitochondrial respiration | ↓ | ↓ |
| Target | Drug | Clinical Significance/Therapeutic Benefits |
|---|---|---|
| PDGF receptor [44,45] | Imatinib | RVSP↓ PAP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ |
| Nilotinib | Terminated due to severe adverse events | |
| PI3K/AKT pathway [47,48] | LY294002 | Cell proliferation↓ |
| Sorafenib | RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ Cell proliferation↓ Cell apoptosis↑ | |
| Rapamycin | PAP↓ RVSP↓ RV hypertrophy↓ Pulmonary vascular remodeling↓ Cell proliferation↓ | |
| Glycolytic metabolism [53,54] | Dichloroacetate | Mitochondrial respiration↑ mPAP↓ PVR↓ |
| miR125a-5p | MCT-induced PASMCs glycolysis↓ MCT-induced PASMCs proliferation↓ RV hypertrophy↓ mPAP↓ | |
| HSP90 [56,57] | 17-AAG | Pulmonary vascular remodeling↓ Cell proliferation↓ Cell migration↓ |
| Gamitrinib | Cell proliferation↓ Cell apoptosis↑ mPAP↓ RVSP↓ Pulmonary vascular remodeling↓ | |
| HMGB1 [58,59] | Saquinavir | Hemodynamic parameters↓ Pulmonary vascular remodeling↓ |
| Glycyrrhizn | Hemodynamic parameters↓ Pulmonary vascular remodeling↓ | |
| BET proteins [61,62] | Apabetalone (RVX208) | Cell proliferation↓ Cell apoptosis↑ Hemodynamics parameters↓ Pulmonary vascular remodeling↓ |
| p53 activation [66] | CP-31398 | HSP90 activity↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, M.-C.W.; Wang, W.-T.; Yeh, J.-L.; Lin, C.-Y.; Kuo, Y.-R.; Lee, S.-S.; Hou, M.-F.; Wu, Y.-C. The Potential Application and Promising Role of Targeted Therapy in Pulmonary Arterial Hypertension. Biomedicines 2022, 10, 1415. https://doi.org/10.3390/biomedicines10061415
Hsieh M-CW, Wang W-T, Yeh J-L, Lin C-Y, Kuo Y-R, Lee S-S, Hou M-F, Wu Y-C. The Potential Application and Promising Role of Targeted Therapy in Pulmonary Arterial Hypertension. Biomedicines. 2022; 10(6):1415. https://doi.org/10.3390/biomedicines10061415
Chicago/Turabian StyleHsieh, Meng-Chien Willie, Wei-Ting Wang, Jwu-Lai Yeh, Chuang-Yu Lin, Yur-Ren Kuo, Su-Shin Lee, Ming-Feng Hou, and Yi-Chia Wu. 2022. "The Potential Application and Promising Role of Targeted Therapy in Pulmonary Arterial Hypertension" Biomedicines 10, no. 6: 1415. https://doi.org/10.3390/biomedicines10061415
APA StyleHsieh, M.-C. W., Wang, W.-T., Yeh, J.-L., Lin, C.-Y., Kuo, Y.-R., Lee, S.-S., Hou, M.-F., & Wu, Y.-C. (2022). The Potential Application and Promising Role of Targeted Therapy in Pulmonary Arterial Hypertension. Biomedicines, 10(6), 1415. https://doi.org/10.3390/biomedicines10061415