
TIRAP/Mal Positively Regulates TLR8-Mediated Signaling via IRF5 in Human Cells

 and
and
Abstract
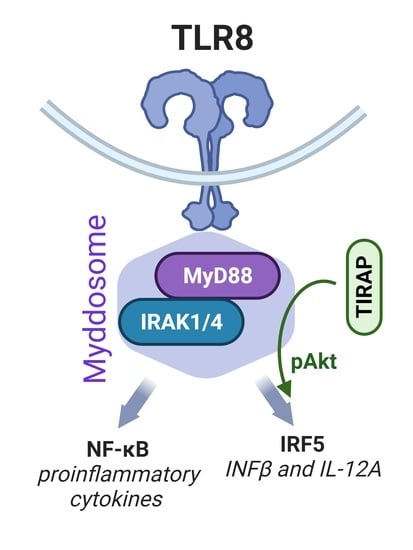
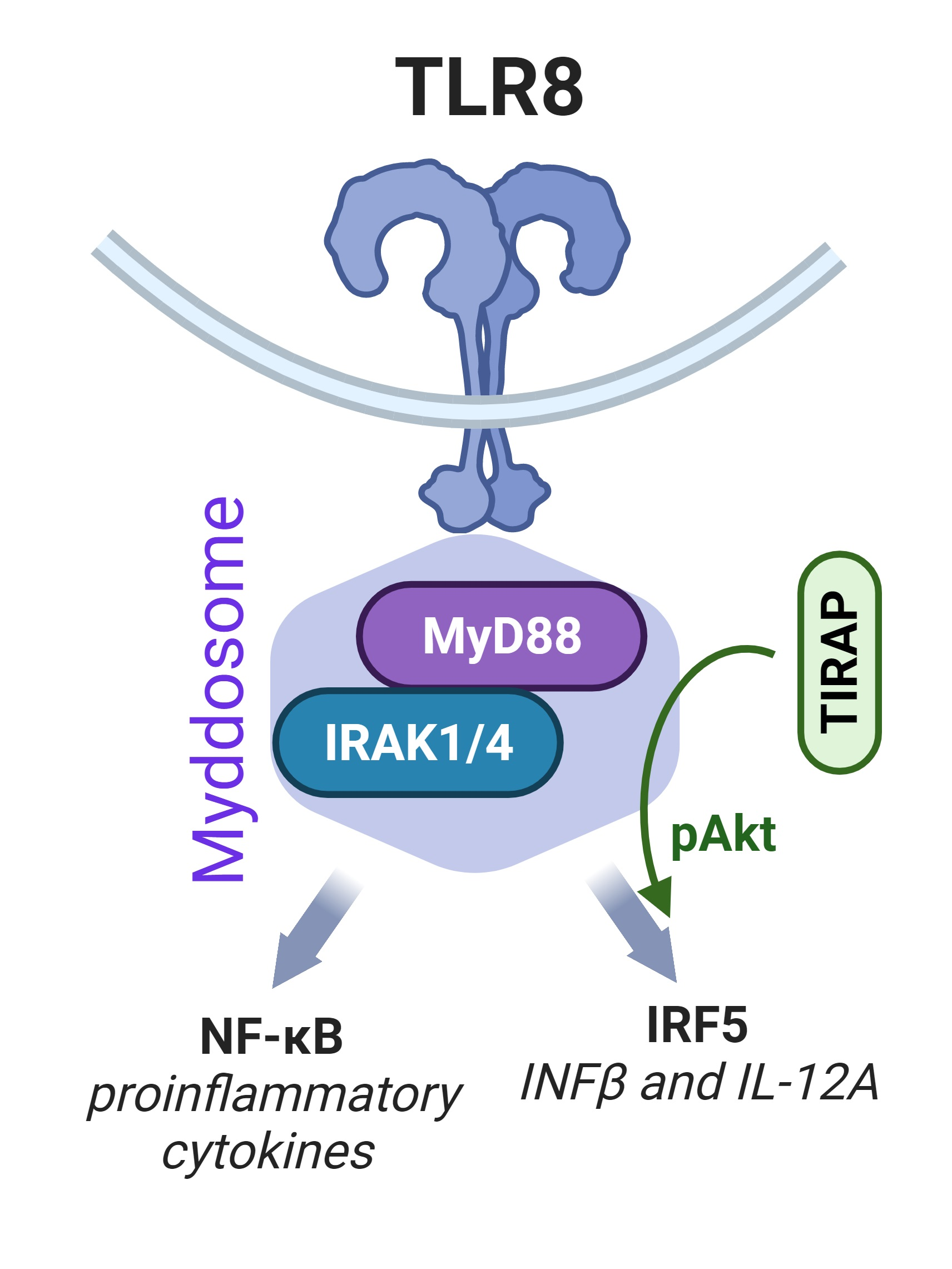
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Antibodies
2.3. siRNA Treatment
2.4. RT-qPCR
2.5. ELISA and BioPlex Assays
2.6. Cell Fractionation
2.7. Western Blotting
2.8. Immunoprecipitations
2.9. Immunofluorescence and Scan^R Analysis
2.10. Bacteria and Infection Experiments
2.11. Statistical Analysis
3. Results
3.1. TIRAP-Silencing Decreases TLR8-Mediated Cytokine Expression and Secretion in Human Primary MDMs
3.2. TIRAP Silencing Inhibits TLR8-Dependent IL-12A Expression in Response to Bacterial Infection
3.3. TLR8-Mediated Nuclear Translocation of IRF5 Is Reduced by TIRAP Silencing
3.4. TIRAP Co-Precipitates with the Myddosome Complex Induced by TLR8 Dimerisation
3.5. TLR8-Mediated Akt Phosphorylation Is Negatively Affected by TIRAP Silencing
3.6. p38 MAPK Inhibition Is Not Affecting TLR8-Mediated IRF5 and p65 Nuclear Translocation
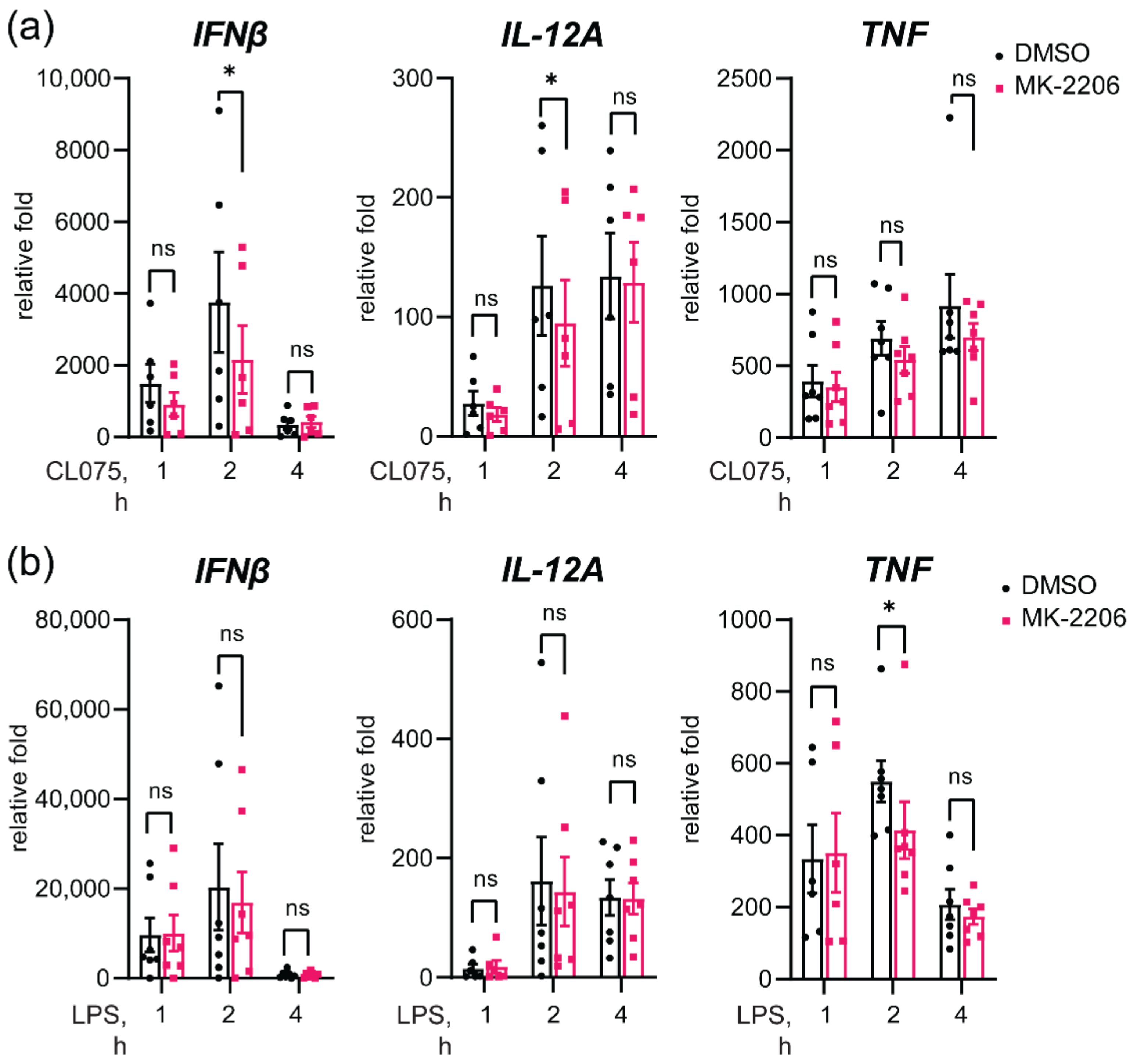
3.7. Akt Inhibition Decreases TLR8-Mediated Expression of IFNβ and IL-12A Genes
3.8. Akt Inhibition Decreases TLR8-Mediated Nuclear Translocation of IRF5
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Sarvestani, S.T.; Williams, B.R.; Gantier, M.P. Human Toll-like receptor 8 can be cool too: Implications for foreign RNA sensing. J. Interferon Cytokine Res. 2012, 32, 350–361. [Google Scholar] [CrossRef]
- Marques, J.T.; Williams, B.R. Activation of the mammalian immune system by siRNAs. Nat. Biotechnol. 2005, 23, 1399–1405. [Google Scholar] [CrossRef]
- Cervantes, J.L.; La Vake, C.J.; Weinerman, B.; Luu, S.; O’Connell, C.; Verardi, P.H.; Salazar, J.C. Human TLR8 is activated upon recognition of Borrelia burgdorferi RNA in the phagosome of human monocytes. J. Leukoc. Biol. 2013, 94, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes, J.L.; Weinerman, B.; Basole, C.; Salazar, J.C. TLR8: The forgotten relative revindicated. Cell. Mol. Immunol. 2012, 9, 434–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, H.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. [Google Scholar] [CrossRef]
- Eigenbrod, T.; Pelka, K.; Latz, E.; Kreikemeyer, B.; Dalpke, A.H. TLR8 Senses Bacterial RNA in Human Monocytes and Plays a Nonredundant Role for Recognition of Streptococcus pyogenes. J. Immunol. 2015, 195, 1092–1099. [Google Scholar] [CrossRef] [Green Version]
- Coch, C.; Hommertgen, B.; Zillinger, T.; Dassler-Plenker, J.; Putschli, B.; Nastaly, M.; Kummerer, B.M.; Scheunemann, J.F.; Schumak, B.; Specht, S.; et al. Human TLR8 Senses RNA From Plasmodium falciparum-Infected Red Blood Cells Which Is Uniquely Required for the IFN-gamma Response in NK Cells. Front. Immunol. 2019, 10, 371. [Google Scholar] [CrossRef] [Green Version]
- Guiducci, C.; Gong, M.; Cepika, A.M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef]
- Greulich, W.; Wagner, M.; Gaidt, M.M.; Stafford, C.; Cheng, Y.; Linder, A.; Carell, T.; Hornung, V. TLR8 Is a Sensor of RNase T2 Degradation Products. Cell 2019, 179, 1264–1275.e1213. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, B.; Aune, M.H.; Awuh, J.A.; Kojen, J.F.; Blix, K.J.; Ryan, L.; Flo, T.H.; Mollnes, T.E.; Espevik, T.; Stenvik, J. TLR8 Senses Staphylococcus aureus RNA in Human Primary Monocytes and Macrophages and Induces IFN-beta Production via a TAK1-IKKbeta-IRF5 Signaling Pathway. J. Immunol. 2015, 195, 1100–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaro, G.; Kostaras, E.; Vivanco, I. Inhibitors in AKTion: ATP-competitive vs allosteric. Biochem. Soc. Trans. 2020, 48, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Ruse, M.; Knaus, U.G. New players in TLR-mediated innate immunity: PI3K and small Rho GTPases. Immunol. Res. 2006, 34, 33–48. [Google Scholar] [CrossRef]
- Aksoy, E.; Vanden Berghe, W.; Detienne, S.; Amraoui, Z.; Fitzgerald, K.A.; Haegeman, G.; Goldman, M.; Willems, F. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-kappa B activation and IFN-beta synthesis downstream of Toll-like receptor 3 and 4. Eur. J. Immunol. 2005, 35, 2200–2209. [Google Scholar] [CrossRef]
- Guiducci, C.; Ghirelli, C.; Marloie-Provost, M.A.; Matray, T.; Coffman, R.L.; Liu, Y.J.; Barrat, F.J.; Soumelis, V. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J. Exp. Med. 2008, 205, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, N.J.; O’Neill, L.A. Mal, more than a bridge to MyD88. IUBMB Life 2013, 65, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.C.; Kagan, J.C. Lipids that directly regulate innate immune signal transduction. Innate Immun. 2020, 26, 4–14. [Google Scholar] [CrossRef]
- Bonham, K.S.; Orzalli, M.H.; Hayashi, K.; Wolf, A.I.; Glanemann, C.; Weninger, W.; Iwasaki, A.; Knipe, D.M.; Kagan, J.C. A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell 2014, 156, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Leszczynska, E.; Makuch, E.; Mitkiewicz, M.; Jasyk, I.; Narita, M.; Gorska, S.; Lipinski, T.; Siednienko, J. Absence of Mal/TIRAP Results in Abrogated Imidazoquinolinones-Dependent Activation of IRF7 and Suppressed IFNbeta and IFN-I Activated Gene Production. Int. J. Mol. Sci. 2020, 21, 8925. [Google Scholar] [CrossRef]
- Zyzak, J.; Mitkiewicz, M.; Leszczynska, E.; Reniewicz, P.; Moynagh, P.N.; Siednienko, J. HSV-1/TLR9-Mediated IFNbeta and TNFalpha Induction Is Mal-Dependent in Macrophages. J. Innate Immun. 2020, 12, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Husebye, H.; Aune, M.H.; Stenvik, J.; Samstad, E.; Skjeldal, F.; Halaas, O.; Nilsen, N.J.; Stenmark, H.; Latz, E.; Lien, E.; et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity 2010, 33, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Ehrnstrom, B.; Beckwith, K.S.; Yurchenko, M.; Moen, S.H.; Kojen, J.F.; Lentini, G.; Teti, G.; Damas, J.K.; Espevik, T.; Stenvik, J. Toll-Like Receptor 8 Is a Major Sensor of Group B Streptococcus But Not Escherichia coli in Human Primary Monocytes and Macrophages. Front. Immunol. 2017, 8, 1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moen, S.H.; Ehrnstrom, B.; Kojen, J.F.; Yurchenko, M.; Beckwith, K.S.; Afset, J.E.; Damas, J.K.; Hu, Z.; Yin, H.; Espevik, T.; et al. Human Toll-like Receptor 8 (TLR8) Is an Important Sensor of Pyogenic Bacteria, and Is Attenuated by Cell Surface TLR Signaling. Front. Immunol. 2019, 10, 1209. [Google Scholar] [CrossRef] [Green Version]
- Horng, T.; Barton, G.M.; Medzhitov, R. TIRAP: An adapter molecule in the Toll signaling pathway. Nat. Immunol. 2001, 2, 835–841. [Google Scholar] [CrossRef]
- Oosenbrug, T.; van de Graaff, M.J.; Haks, M.C.; van Kasteren, S.; Ressing, M.E. An alternative model for type I interferon induction downstream of human TLR2. J. Biol. Chem. 2020, 295, 14325–14342. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurchenko, M.; Skjesol, A.; Ryan, L.; Richard, G.M.; Kandasamy, R.K.; Wang, N.; Terhorst, C.; Husebye, H.; Espevik, T. SLAMF1 is required for TLR4-mediated TRAM-TRIF-dependent signaling in human macrophages. J. Cell Biol. 2018, 217, 1411–1429. [Google Scholar] [CrossRef]
- Anwar, S.; Prince, L.R.; Foster, S.J.; Whyte, M.K.; Sabroe, I. The rise and rise of Staphylococcus aureus: Laughing in the face of granulocytes. Clin. Exp. Immunol. 2009, 157, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcaite, E.; Bartusevicius, A.; Tameliene, R.; Kliucinskas, M.; Maleckiene, L.; Nadisauskiene, R. Prevalence of maternal group B streptococcal colonisation in European countries. Acta Obstet. Gynecol. Scand. 2008, 87, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pelaez, M.; Lamont, D.J.; Peggie, M.; Shpiro, N.; Gray, N.S.; Cohen, P. Protein kinase IKKbeta-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 17432–17437. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. IRF and STAT Transcription Factors—From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies. Front. Immunol. 2018, 9, 3047. [Google Scholar] [CrossRef]
- Nagy, Z.; Comer, S.; Smolenski, A. Analysis of Protein Phosphorylation Using Phos-Tag Gels. Curr. Protoc. Protein Sci. 2018, 93, e64. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Horng, T.; Barton, G.M.; Flavell, R.A.; Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002, 420, 329–333. [Google Scholar] [CrossRef]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, S.; Strickson, S.; Zhang, T.; Gray, N.; Lee, K.L.; Rao, V.R.; Cohen, P. The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and Toll-like receptor agonists. Biochem. J. 2017, 474, 2027–2038. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Troutman, T.D.; Bazan, J.F.; Pasare, C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle 2012, 11, 3559–3567. [Google Scholar] [CrossRef] [Green Version]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belhaouane, I.; Hoffmann, E.; Chamaillard, M.; Brodin, P.; Machelart, A. Paradoxical Roles of the MAL/Tirap Adaptor in Pathologies. Front. Immunol. 2020, 11, 569127. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C. Defining the subcellular sites of innate immune signal transduction. Trends Immunol. 2012, 33, 442–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, L.; Wang, Y.; Bulek, K.; Della Mina, E.; Zhang, Z.; Pedergnana, V.; Chrabieh, M.; Lemmens, N.A.; Sancho-Shimizu, V.; Descatoire, M.; et al. Human Adaptive Immunity Rescues an Inborn Error of Innate Immunity. Cell 2017, 168, 789–800.e710. [Google Scholar] [CrossRef] [Green Version]
- Troutman, T.D.; Hu, W.; Fulenchek, S.; Yamazaki, T.; Kurosaki, T.; Bazan, J.F.; Pasare, C. Role for B-cell adapter for PI3K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3K/Akt. Proc. Natl. Acad. Sci. USA 2012, 109, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Hamerman, J.A.; Pottle, J.; Ni, M.; He, Y.; Zhang, Z.Y.; Buckner, J.H. Negative regulation of TLR signaling in myeloid cells--implications for autoimmune diseases. Immunol. Rev. 2016, 269, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Lima, B.H.F.; Marques, P.E.; Gomides, L.F.; Mattos, M.S.; Kraemer, L.; Queiroz-Junior, C.M.; Lennon, M.; Hirsch, E.; Russo, R.C.; Menezes, G.B.; et al. Converging TLR9 and PI3Kgamma signaling induces sterile inflammation and organ damage. Sci. Rep. 2019, 9, 19085. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.N.; Peters, K.L.; Elco, C.P.; Sakamoto, S.; Pal, S.; Sen, G.C. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol. 2004, 11, 1060–1067. [Google Scholar] [CrossRef]
- Heinz, L.X.; Lee, J.; Kapoor, U.; Kartnig, F.; Sedlyarov, V.; Papakostas, K.; Cesar-Razquin, A.; Essletzbichler, P.; Goldmann, U.; Stefanovic, A.; et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature 2020, 581, 316–322. [Google Scholar] [CrossRef]
- Sartorius, R.; Trovato, M.; Manco, R.; D’Apice, L.; De Berardinis, P. Exploiting viral sensing mediated by Toll-like receptors to design innovative vaccines. NPJ Vaccines 2021, 6, 127. [Google Scholar] [CrossRef]
- Martinez-Espinoza, I.; Guerrero-Plata, A. The Relevance of TLR8 in Viral Infections. Pathogens 2022, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- de Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal 2019, 12, eaaw1347. [Google Scholar] [CrossRef] [PubMed]
- Thada, S.; Horvath, G.L.; Muller, M.M.; Dittrich, N.; Conrad, M.L.; Sur, S.; Hussain, A.; Pelka, K.; Gaddam, S.L.; Latz, E.; et al. Interaction of TLR4 and TLR8 in the Innate Immune Response against Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2021, 22, 1560. [Google Scholar] [CrossRef] [PubMed]
- Sakaniwa, K.; Shimizu, T. Targeting the innate immune receptor TLR8 using small-molecule agents. Acta Crystallogr. D Struct. Biol. 2020, 76, 621–629. [Google Scholar] [CrossRef]
- Meas, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbo, S.A.; Tasken, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 activates human T cells and reverses latency. Nat. Commun. 2020, 11, 147. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsen, K.E.; Skjesol, A.; Frengen Kojen, J.; Espevik, T.; Stenvik, J.; Yurchenko, M. TIRAP/Mal Positively Regulates TLR8-Mediated Signaling via IRF5 in Human Cells. Biomedicines 2022, 10, 1476. https://doi.org/10.3390/biomedicines10071476
Nilsen KE, Skjesol A, Frengen Kojen J, Espevik T, Stenvik J, Yurchenko M. TIRAP/Mal Positively Regulates TLR8-Mediated Signaling via IRF5 in Human Cells. Biomedicines. 2022; 10(7):1476. https://doi.org/10.3390/biomedicines10071476
Chicago/Turabian StyleNilsen, Kaja Elisabeth, Astrid Skjesol, June Frengen Kojen, Terje Espevik, Jørgen Stenvik, and Maria Yurchenko. 2022. "TIRAP/Mal Positively Regulates TLR8-Mediated Signaling via IRF5 in Human Cells" Biomedicines 10, no. 7: 1476. https://doi.org/10.3390/biomedicines10071476