
Novel Inflammasome-Based Risk Score for Predicting Survival and Efficacy to Immunotherapy in Early-Stage Non-Small Cell Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
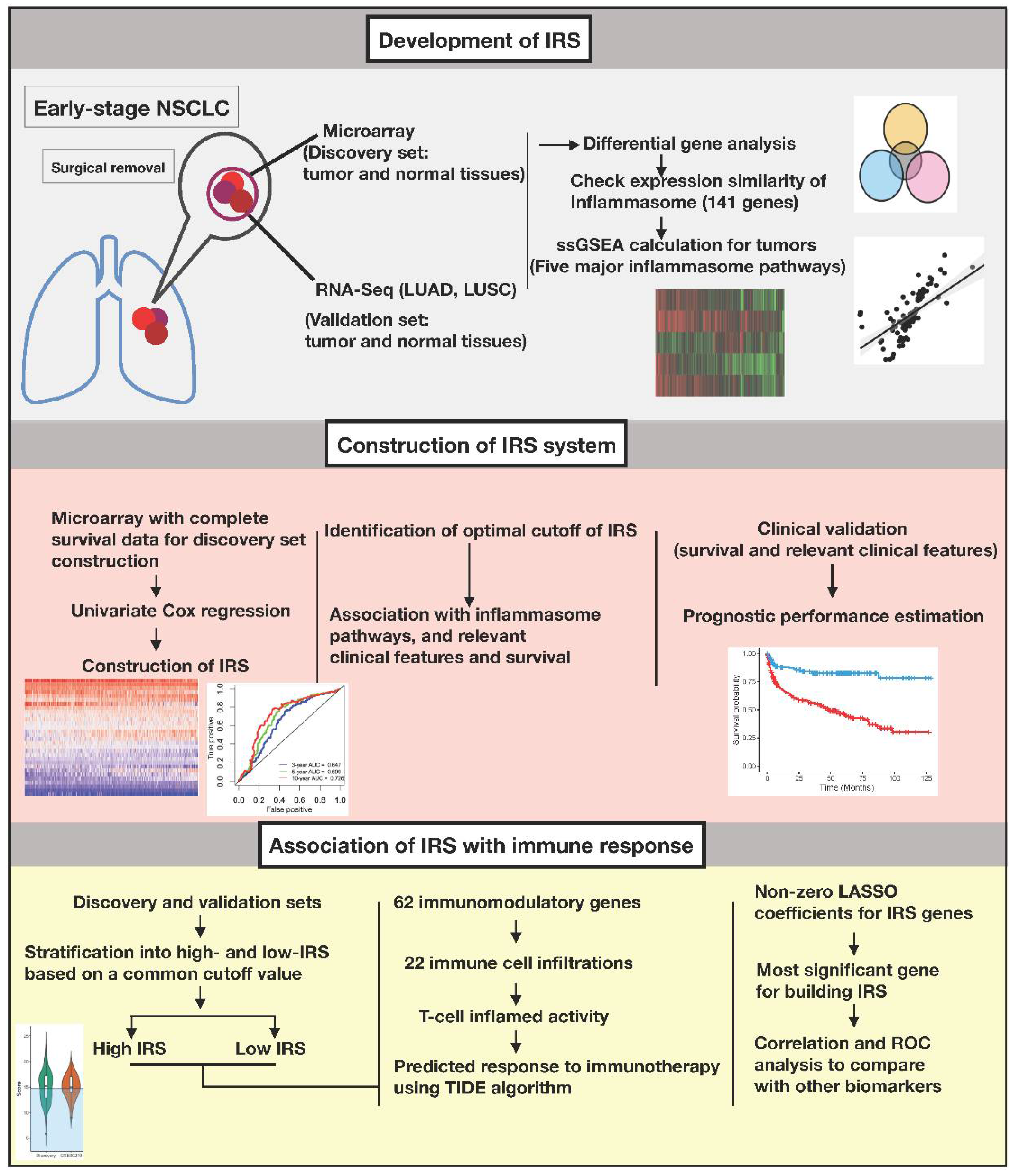
2. Materials and Methods
2.1. Microarray Data
2.2. RNA-Seq Data
2.3. Construction of the Inflammasome Signature
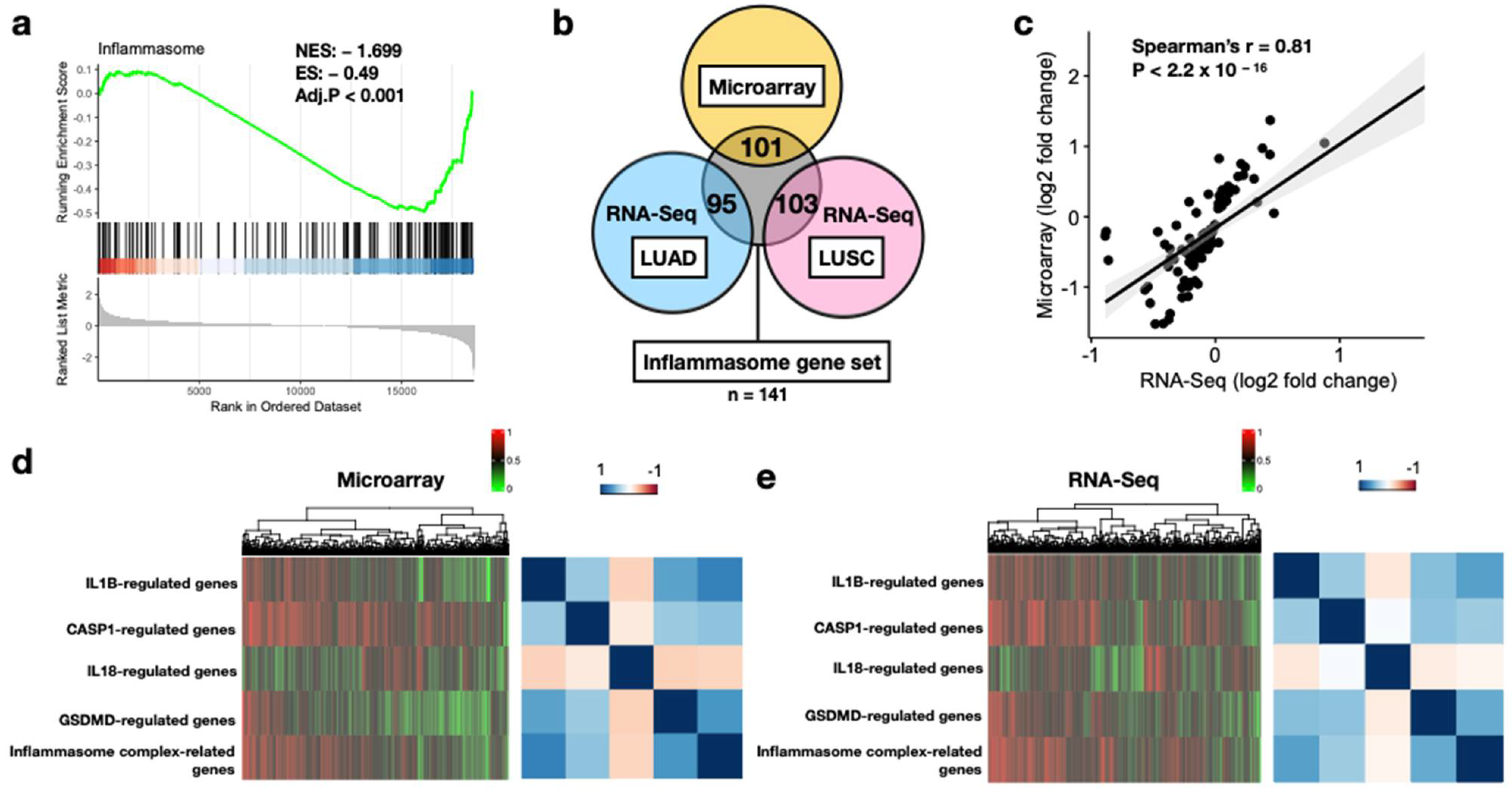
2.4. Gene Set Enrichment Analysis (GSEA) and Differential Gene Analysis
2.5. Construction of IRS
2.6. Prognostic Value of IRS
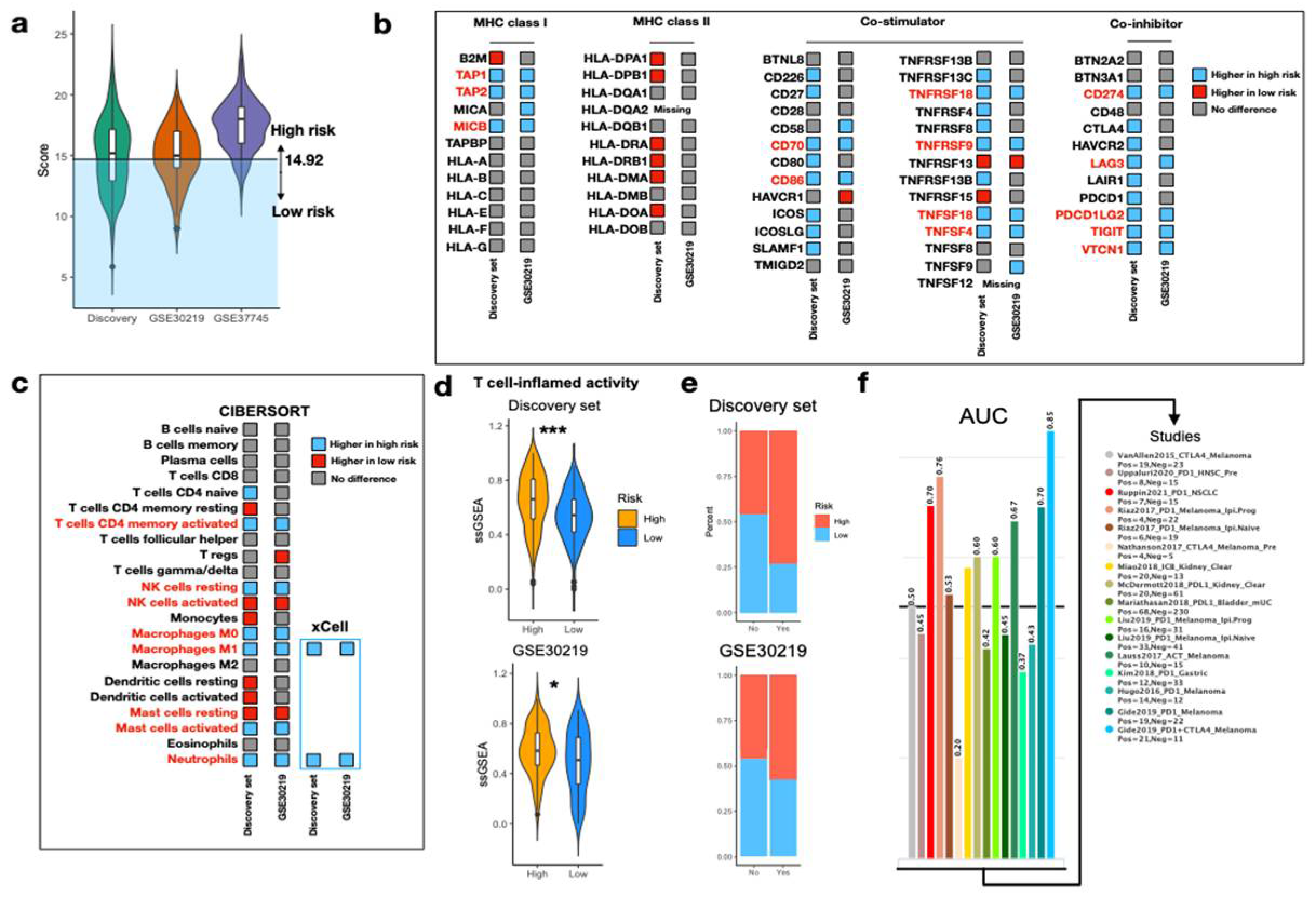
2.7. Estimation of Immune Response
2.8. Survival Analysis
2.9. Statistical Analysis
3. Results
3.1. Feasibility of Core Inflammasome Genes in Early-Stage NSCLC
3.2. Construction of a 30-Gene IRS
3.3. IRS Is Associated with Inflammasome Activity and Various Clinical Features in NSCLC
3.4. Prognostic Potential of IRS
3.5. Predict Response to ICI via IRS
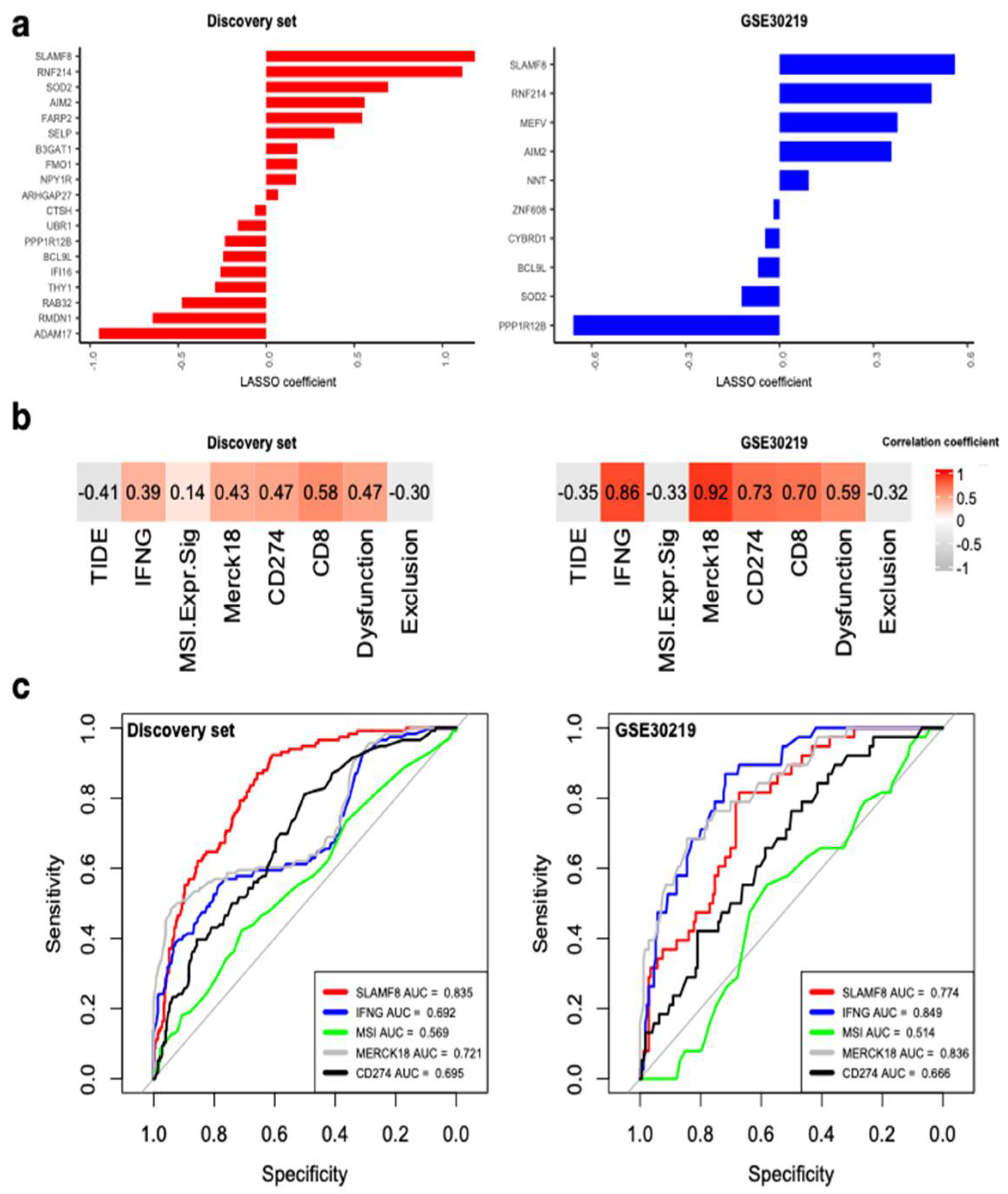
3.6. SLAMF8 Is the Key Inflammasome Gene for Predicting ICI Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gerber, D.E.; Oxnard, G.R.; Govindan, R. ALCHEMIST: Bringing genomic discovery and targeted therapies to early-stage lung cancer. Clin. Pharm. 2015, 97, 447–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, M.; Herman, J.G. Noninvasive Diagnostics for Early Detection of Lung Cancer: Challenges and Potential with a Focus on Changes in DNA Methylation. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Uramoto, H.; Tanaka, F. Recurrence after surgery in patients with NSCLC. Transl. Lung Cancer Res. 2014, 3, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Gaspar, L.E.; Chaft, J.E.; Kennedy, E.B.; Azzoli, C.G.; Ellis, P.M.; Lin, S.H.; Pass, H.I.; Seth, R.; Shepherd, F.A.; et al. Adjuvant Systemic Therapy and Adjuvant Radiation Therapy for Stage I to IIIA Completely Resected Non-Small-Cell Lung Cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 2960–2974. [Google Scholar] [CrossRef] [Green Version]
- Pignon, J.P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L.; et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Yeh, J.; Marrone, K.A.; Forde, P.M. Neoadjuvant and consolidation immuno-oncology therapy in stage III non-small cell lung cancer. J. Thorac. Dis. 2018, 10, S451–S459. [Google Scholar] [CrossRef] [Green Version]
- Felip, E.; Altorki, N.; Zhou, C.; Csoszi, T.; Vynnychenko, I.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H.; et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase 3 trial. Lancet 2021, 398, 1344–1357. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, F.; Klotz, L.V.; Bischoff, H.; Thomas, M.; Lasitschka, F.; Winter, H.; Hoffmann, H.; Eichhorn, M.E. Neoadjuvant anti-programmed Death-1 immunotherapy by Pembrolizumab in resectable nodal positive stage II/IIIa non-small-cell lung cancer (NSCLC): The NEOMUN trial. BMC Cancer 2019, 19, 413. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Weinberg, R.A. Tumor-host interactions: A far-reaching relationship. J. Clin. Oncol. 2010, 28, 4022–4028. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, P.; Vora, S.M.; Wu, H. Higher-order assemblies in innate immune and inflammatory signaling: A general principle in cell biology. Curr. Opin. Cell Biol. 2020, 63, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Li, X.Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef] [Green Version]
- Segovia, M.; Russo, S.; Jeldres, M.; Mahmoud, Y.D.; Perez, V.; Duhalde, M.; Charnet, P.; Rousset, M.; Victoria, S.; Veigas, F.; et al. Targeting TMEM176B Enhances Antitumor Immunity and Augments the Efficacy of Immune Checkpoint Blockers by Unleashing Inflammasome Activation. Cancer Cell 2019, 35, 767–781.e766. [Google Scholar] [CrossRef] [Green Version]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Wu, J.; Zhao, X.; Shen, S.; Zhu, C.; Liu, T.; Cui, X.; Chen, L.; Wei, C.; Cheng, P.; et al. Establishment of tumor inflammasome clusters with distinct immunogenomic landscape aids immunotherapy. Theranostics 2021, 11, 9884–9903. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Nanayakkara, G.; Sun, Y.; Li, X.; Wang, L.; Cueto, R.; Shao, Y.; Fu, H.; Johnson, C.; Cheng, J.; et al. Analyses of caspase-1-regulated transcriptomes in various tissues lead to identification of novel IL-1beta-, IL-18- and sirtuin-1-independent pathways. J. Hematol. Oncol. 2017, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Indini, A.; Rijavec, E.; Bareggi, C.; Grossi, F. Novel treatment strategies for early-stage lung cancer: The oncologist’s perspective. J. Thorac. Dis. 2020, 12, 3390–3398. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Zhao, J.; Xing, Y.; Zhang, X.; Liu, J.; Ouyang, Q.; Chen, J.; Su, F.; Liu, Q.; Song, E. Immune Checkpoint Inhibition Overcomes ADCP-Induced Immunosuppression by Macrophages. Cell 2018, 175, 442–457.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Wang, Y.; Zeng, X.; Wang, Z.; Wang, H.; Xie, W. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumor Biol. 2015, 36, 7501–7513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jin, C.; Yang, Y.; Wang, K.; Zhou, Y.; Zhou, Y.; Wang, R.; Li, T.; Hu, R. AIM2 promotes non-small-cell lung cancer cell growth through inflammasome-dependent pathway. J. Cell Physiol. 2019, 234, 20161–20173. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Garon, E.B.; Chih-Hsin Yang, J.; Dubinett, S.M. The Role of Interleukin 1beta in the Pathogenesis of Lung Cancer. JTO Clin. Res. Rep. 2020, 1, 100001. [Google Scholar] [CrossRef]
- Yun, J.K.; Lee, G.D.; Kim, H.R.; Kim, Y.H.; Kim, D.K.; Park, S.I.; Choi, S. Validation of the 8th edition of the TNM staging system in 3950 patients with surgically resected non-small cell lung cancer. J. Thorac. Dis. 2019, 11, 2955–2964. [Google Scholar] [CrossRef]
- Goldstraw, P.; Crowley, J.; Chansky, K.; Giroux, D.J.; Groome, P.A.; Rami-Porta, R.; Postmus, P.E.; Rusch, V.; Sobin, L.; International Association for the Study of Lung Cancer International Staging Committee; et al. The IASLC Lung Cancer Staging Project: Proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J. Thorac. Oncol. 2007, 2, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Butcher, M.J.; Galkina, E.V. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front. Physiol. 2012, 3, 44. [Google Scholar] [CrossRef] [Green Version]
- Pelegrin, P.; Surprenant, A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J. 2009, 28, 2114–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wei, Y.; Tang, Z.; Liu, B.; Dong, J. Tumorassociated macrophages in lung cancer: Friend or foe? (Review). Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 Macrophages Induce PD-L1 Expression in Hepatocellular Carcinoma Cells Through IL-1beta Signaling. Front. Immunol. 2019, 10, 1643. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- John, P.; Wei, Y.; Liu, W.; Du, M.; Guan, F.; Zang, X. The B7x Immune Checkpoint Pathway: From Discovery to Clinical Trial. Trends Pharm. Sci. 2019, 40, 883–896. [Google Scholar] [CrossRef]
- Harjunpaa, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef]
- Kim, H.; Chung, J.H. PD-L1 Testing in Non-small Cell Lung Cancer: Past, Present, and Future. J. Pathol. Transl. Med. 2019, 53, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Long-Mira, E.; Bence, C.; Butori, C.; Lassalle, S.; Bouhlel, L.; Fazzalari, L.; Zahaf, K.; Lalvee, S.; Washetine, K.; et al. Comparative study of the PD-L1 status between surgically resected specimens and matched biopsies of NSCLC patients reveal major discordances: A potential issue for anti-PD-L1 therapeutic strategies. Ann. Oncol. 2016, 27, 147–153. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ford, J.M. Multigene Panel Testing in Oncology Practice: How Should We Respond? JAMA Oncol. 2015, 1, 277–278. [Google Scholar] [CrossRef] [Green Version]
- Detre, C.; Keszei, M.; Romero, X.; Tsokos, G.C.; Terhorst, C. SLAM family receptors and the SLAM-associated protein (SAP) modulate T cell functions. Semin. Immunopathol. 2010, 32, 157–171. [Google Scholar] [CrossRef] [Green Version]
- De Calisto, J.; Wang, N.; Wang, G.; Yigit, B.; Engel, P.; Terhorst, C. SAP-Dependent and -Independent Regulation of Innate T Cell Development Involving SLAMF Receptors. Front. Immunol. 2014, 5, 186. [Google Scholar] [CrossRef] [Green Version]
- Romero, X.; Sintes, J.; Engel, P. Role of SLAM family receptors and specific adapter SAP in innate-like lymphocytes. Crit. Rev. Immunol. 2014, 34, 263–299. [Google Scholar] [CrossRef]
- Zhang, Q.; Cheng, L.; Qin, Y.; Kong, L.; Shi, X.; Hu, J.; Li, L.; Ding, Z.; Wang, T.; Shen, J.; et al. SLAMF8 expression predicts the efficacy of anti-PD1 immunotherapy in gastrointestinal cancers. Clin. Transl. Immunol. 2021, 10, e1347. [Google Scholar] [CrossRef]
- Zou, C.Y.; Guan, G.F.; Zhu, C.; Liu, T.Q.; Guo, Q.; Cheng, W.; Wu, A.H. Costimulatory checkpoint SLAMF8 is an independent prognosis factor in glioma. CNS Neurosci. Ther. 2019, 25, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, K.; Gill, R.R.; Widick, P.; Bindal, P.; McDonald, D.C.; Shea, M.; Rangachari, D.; Costa, D.B. Association of Performance Status With Survival in Patients With Advanced Non-Small Cell Lung Cancer Treated With Pembrolizumab Monotherapy. JAMA Netw. Open 2021, 4, e2037120. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsao, C.-C.; Wu, H.-H.; Wang, Y.-F.; Shen, P.-C.; Wu, W.-T.; Chen, H.-Y.; Dai, Y.-H. Novel Inflammasome-Based Risk Score for Predicting Survival and Efficacy to Immunotherapy in Early-Stage Non-Small Cell Lung Cancer. Biomedicines 2022, 10, 1539. https://doi.org/10.3390/biomedicines10071539
Tsao C-C, Wu H-H, Wang Y-F, Shen P-C, Wu W-T, Chen H-Y, Dai Y-H. Novel Inflammasome-Based Risk Score for Predicting Survival and Efficacy to Immunotherapy in Early-Stage Non-Small Cell Lung Cancer. Biomedicines. 2022; 10(7):1539. https://doi.org/10.3390/biomedicines10071539
Chicago/Turabian StyleTsao, Chih-Cheng, Hsin-Hung Wu, Ying-Fu Wang, Po-Chien Shen, Wen-Ting Wu, Huang-Yun Chen, and Yang-Hong Dai. 2022. "Novel Inflammasome-Based Risk Score for Predicting Survival and Efficacy to Immunotherapy in Early-Stage Non-Small Cell Lung Cancer" Biomedicines 10, no. 7: 1539. https://doi.org/10.3390/biomedicines10071539
APA StyleTsao, C.-C., Wu, H.-H., Wang, Y.-F., Shen, P.-C., Wu, W.-T., Chen, H.-Y., & Dai, Y.-H. (2022). Novel Inflammasome-Based Risk Score for Predicting Survival and Efficacy to Immunotherapy in Early-Stage Non-Small Cell Lung Cancer. Biomedicines, 10(7), 1539. https://doi.org/10.3390/biomedicines10071539