
Dirty Jobs: Macrophages at the Heart of Cardiovascular Disease

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
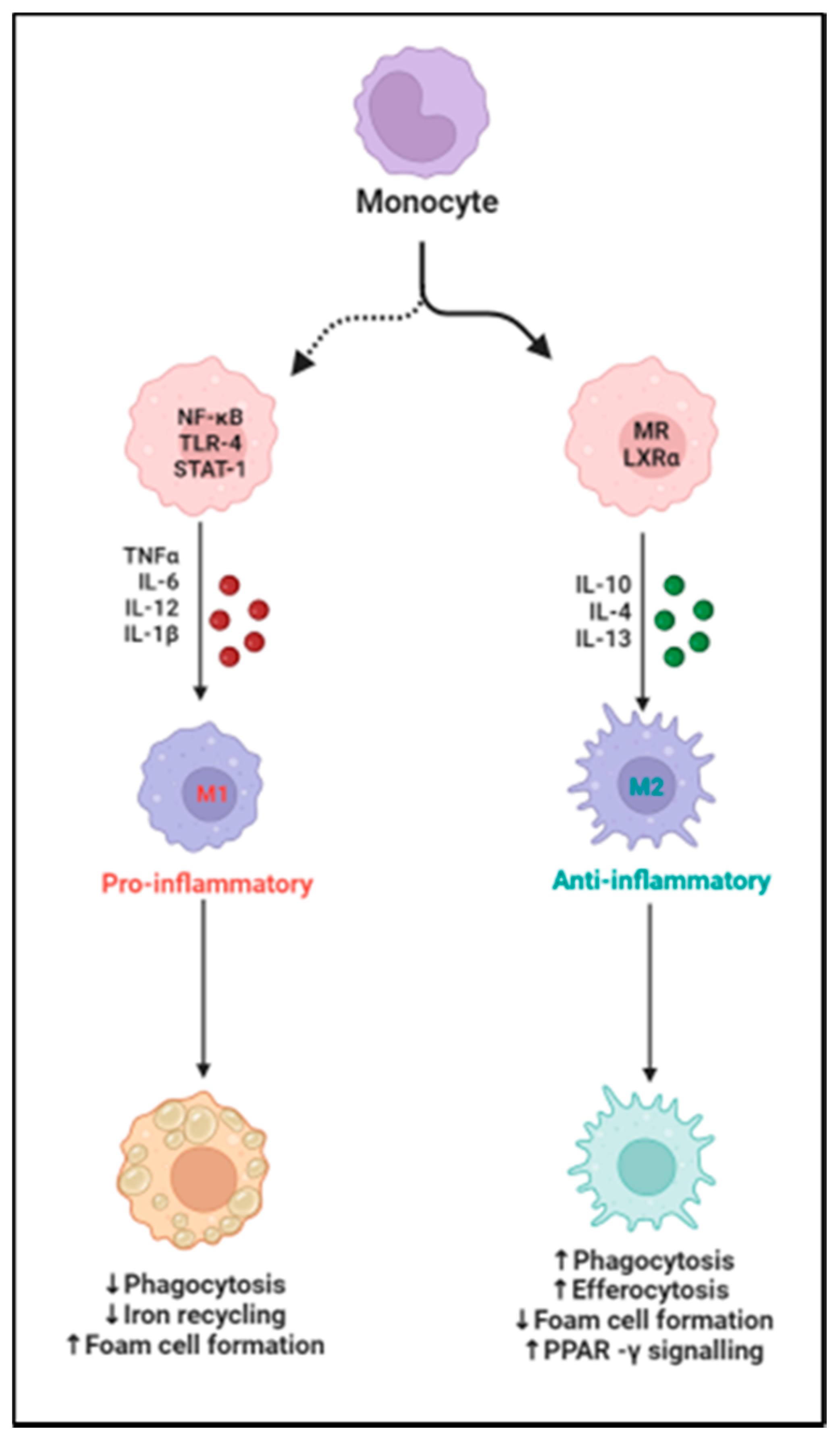
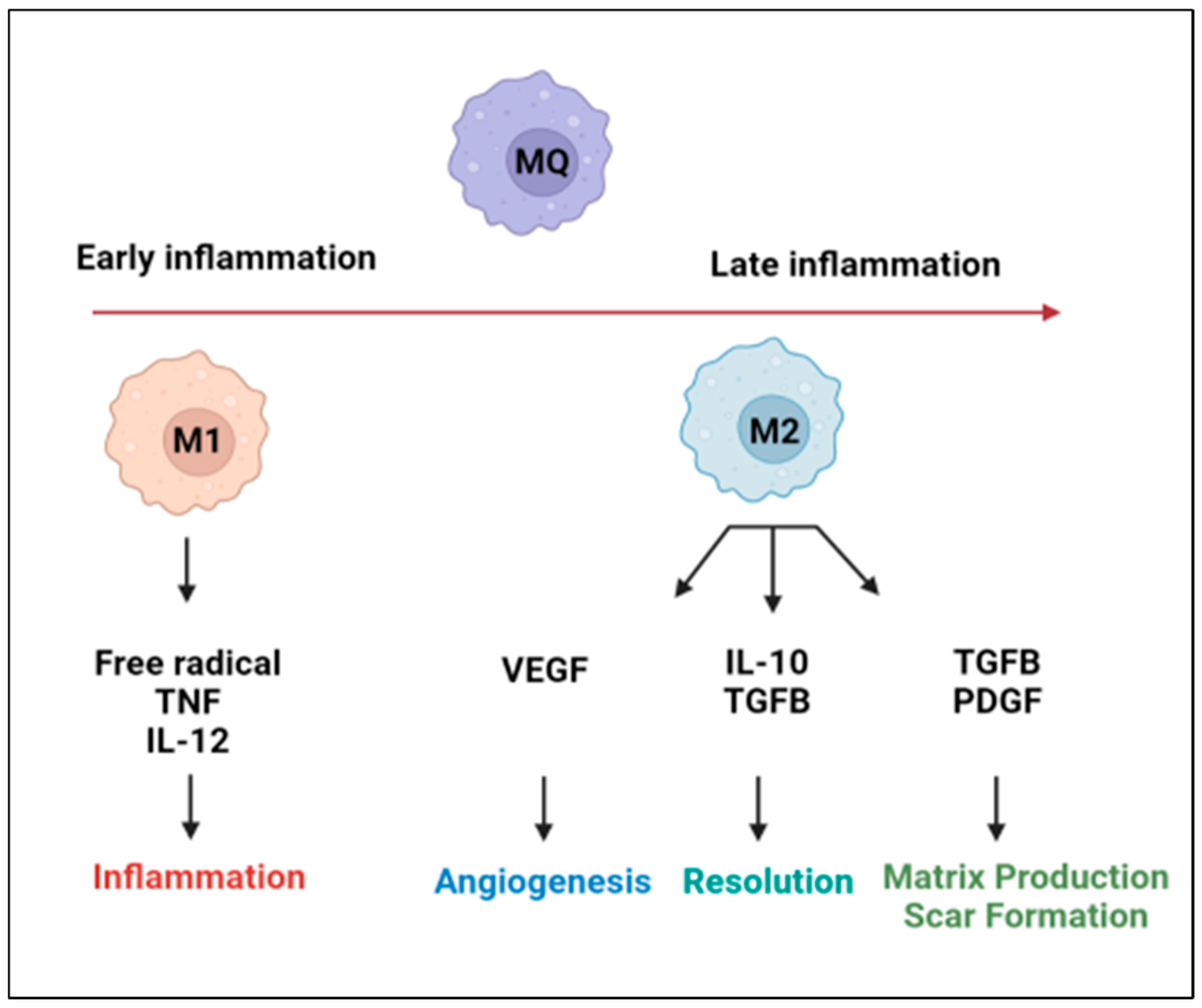
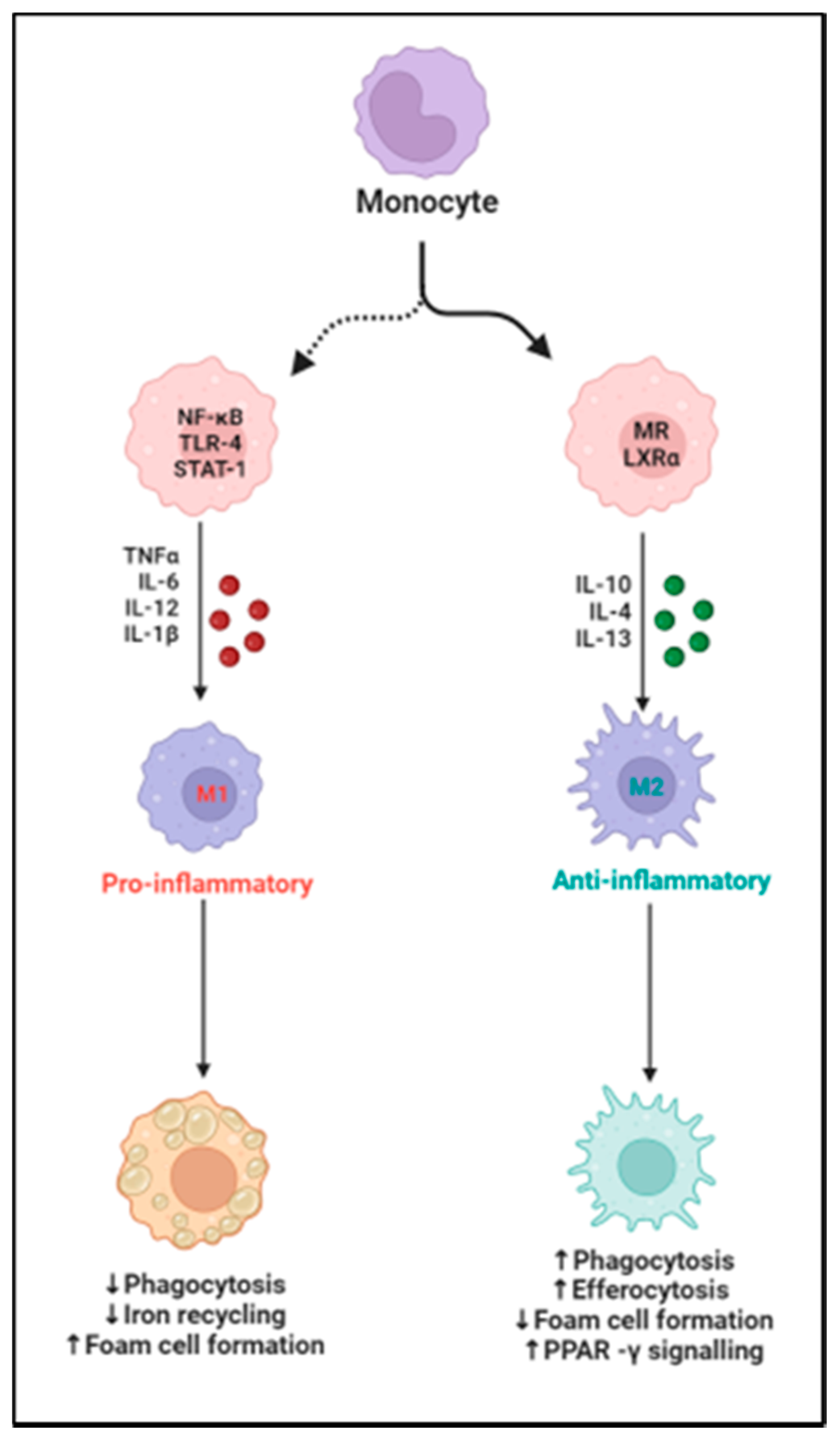
2. Cardiac Macrophage Plasticity
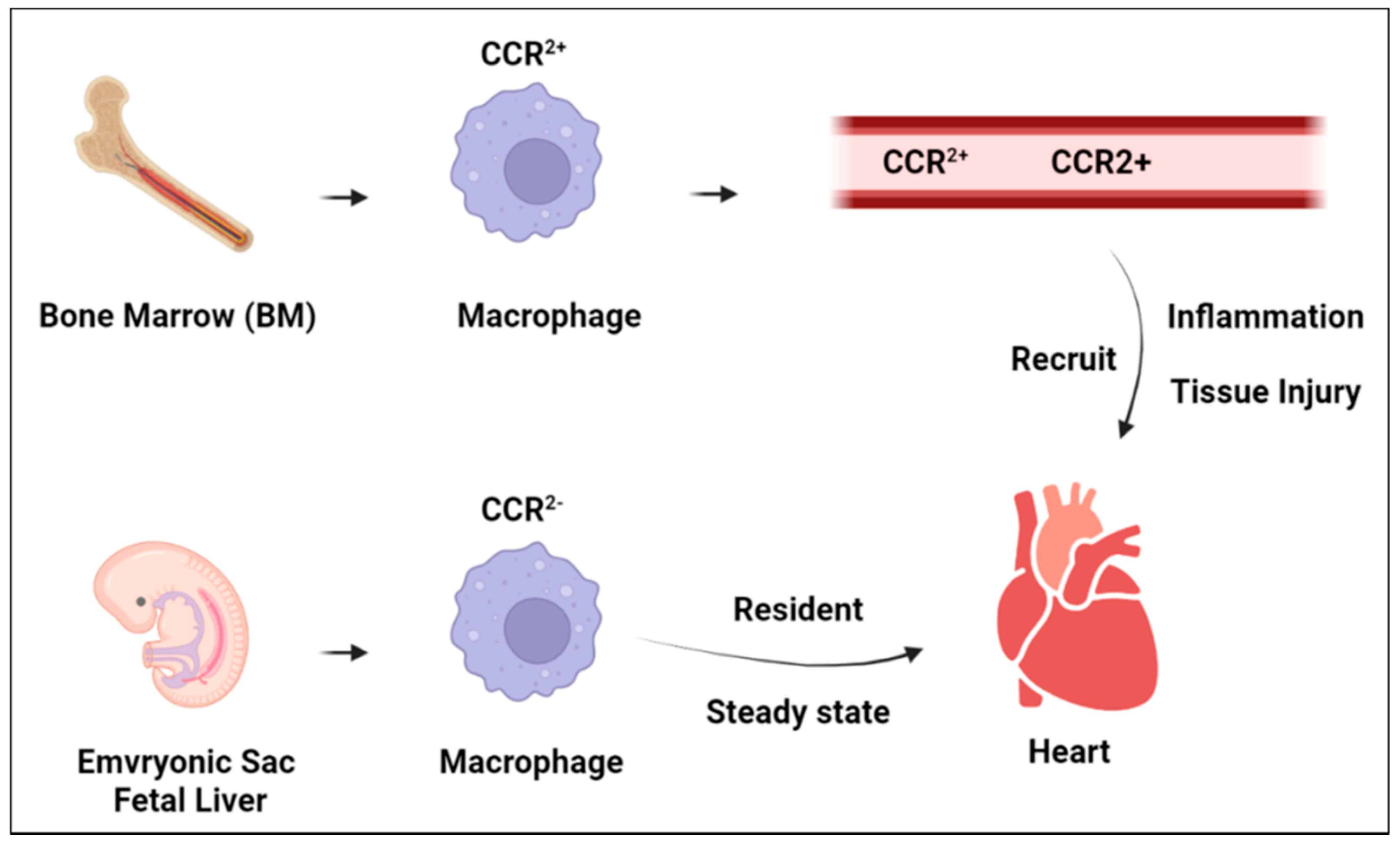
3. Macrophage Function in Heart Homeostasis
4. Macrophages and Heart Failure
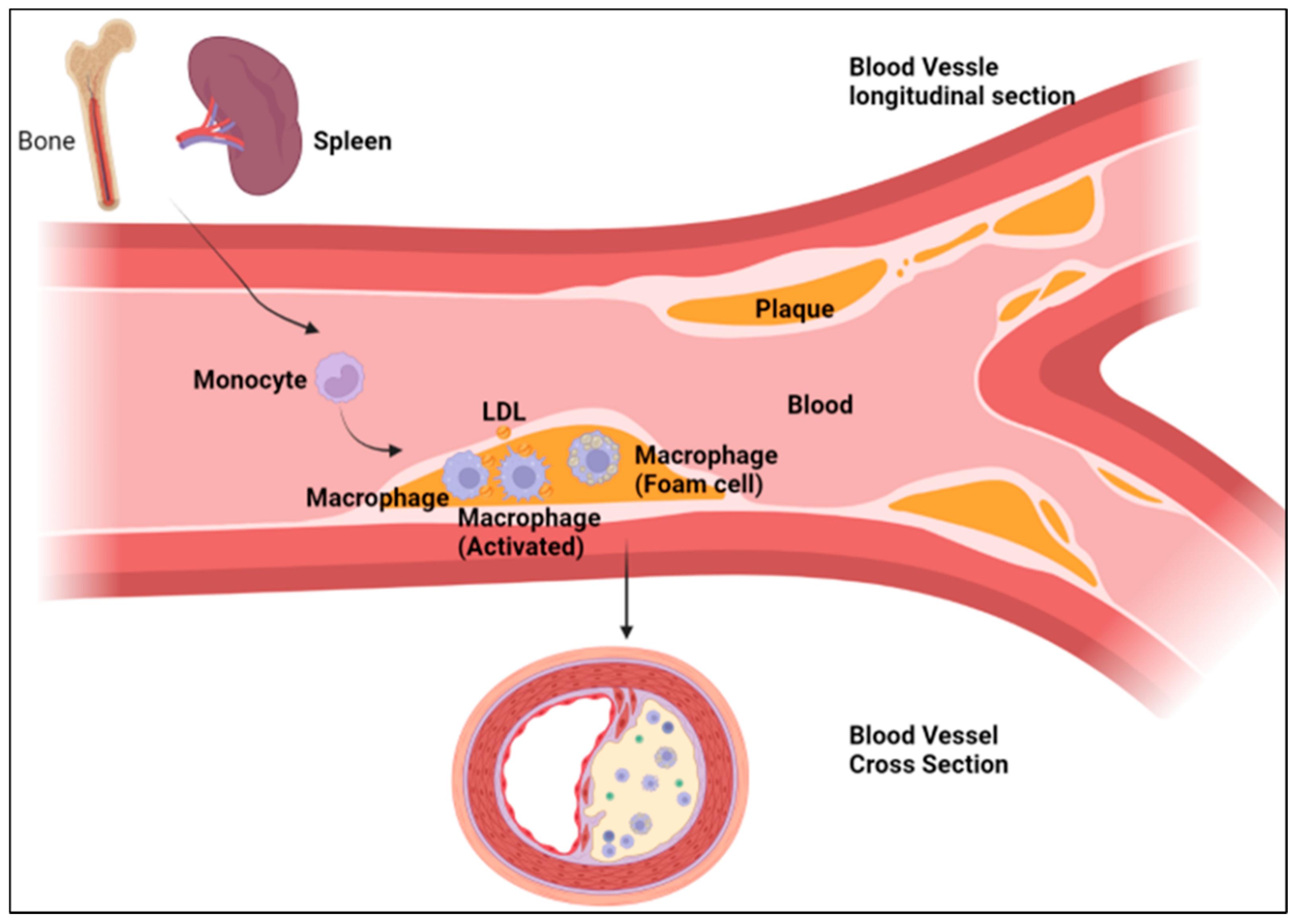
5. Macrophages and Atherosclerosis
6. Macrophages and Cardiomyopathy
7. Targeting Macrophages in the Treatment of CVD
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kddcup1999. 1991. Available online: http://kdd.ics.uci.edu/databases/ (accessed on 28 October 1999).
- Bhatnagar, P.; Wickramasinghe, K.; Williams, J.; Rayner, M.; Townsend, N. The epidemiology of cardiovascular disease in the UK 2014. Heart 2015, 101, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Liu, J.; Wang, M.; Zhang, X.; Zhou, M. Epidemiology of cardiovascular disease in China: Current features and implications. Nat. Rev. Cardiol. 2019, 16, 203–212. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared risk factors in cardiovascular disease and cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, X.-C.; Lu, L.; Cao, Y.; Sun, R.-R.; Chen, S.; Zhang, P.-Y. Cardiovascular disease and its relationship with chronic kidney disease. Eur. Rev. Med. Pharm. Sci. 2014, 18, 2918–2926. [Google Scholar]
- Dube, P.; DeRiso, A.; Patel, M.; Battepati, D.; Khatib-Shahidi, B.; Sharma, H.; Gupta, R.; Malhotra, D.; Dworkin, L.; Haller, S.; et al. Vascular calcification in chronic kidney disease: Diversity in the vessel wall. Biomedicines 2021, 9, 404. [Google Scholar] [CrossRef]
- Lioufas, N.M.; Pedagogos, E.; Hawley, C.M.; Pascoe, E.M.; Elder, G.J.; Badve, S.V.; Valks, A.; Toussaint, N.D. Aortic calcification and arterial stiffness burden in a chronic kidney disease cohort with high cardiovascular risk: Baseline characteristics of the impact of phosphate reduction on vascular end-points in chronic kidney disease trial. Am. J. Nephrol. 2020, 51, 201–215. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Wang, Z.; Koenig, A.L.; Lavine, K.J.; Apte, R.S. Macrophage plasticity and function in the eye and heart. Trends Immunol. 2019, 40, 825–841. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Davies, L.C.T. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.W.; LaVine, K.J. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Moore, K.J. Defining macrophages in the heart one cell at a time. Trends Immunol. 2019, 40, 179–181. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Hidalgo, A.; Ballesteros, I. Specialized functions of resident macrophages in brain and heart. J. Leukoc. Biol. 2018, 104, 743–756. [Google Scholar] [CrossRef]
- Li, W.; Hsiao, H.M.; Higashikubo, R.; Saunders, B.T.; Bharat, A.; Goldstein, D.R.; Krupnick, A.S.; Gelman, A.E.; Lavine, K.J.; Kreisel, D. Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016, 1, e87315. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Yap, J.; Cabrera-Fuentes, H.A.; Irei, J.; Hausenloy, D.J.; Boisvert, W.A. Role of macrophages in cardioprotection. Int. J. Mol. Sci. 2019, 20, 2474. [Google Scholar] [CrossRef] [Green Version]
- Fujiu, K.; Wang, J.; Nagai, R. Cardioprotective function of cardiac macrophages. Cardiovasc. Res. 2014, 102, 232–239. [Google Scholar] [CrossRef]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.R.; Mohan, J.; Kopecky, B.J.; Guo, S.; Du, L.; Leid, J.; Feng, G.; Lokshina, I.; Dmytrenko, O.; Luehmann, H.; et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 2021, 54, 2072–2088.e7. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; AlThagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Aguirre, G.A.; González-Guerra, J.L.; Espinosa, L.; Castilla-Cortazar, I. Insulin-like growth factor 1 in the cardiovascular system. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 1–45. [Google Scholar]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages facilitate electrical conduction in the heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [Green Version]
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef]
- Mudd, J.O.; Kass, D.A. Tackling heart failure in the twenty-first century. Nature 2008, 451, 919–928. [Google Scholar] [CrossRef]
- Kemp, C.D.; Conte, J.V. The pathophysiology of heart failure. Cardiovasc. Pathol. 2012, 21, 365–371. [Google Scholar] [CrossRef]
- Rossignol, P.; Hernandez, A.F.; Solomon, S.D.; Zannad, F. Heart failure drug treatment. Lancet 2019, 393, 1034–1044. [Google Scholar] [CrossRef]
- Moskalik, A.; Niderla-Bielińska, J.; Ratajska, A. Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail. Rev. 2021, 27, 1413–1430. [Google Scholar] [CrossRef]
- McMurray, J.J.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar]
- Schiattarella, G.G.; Tong, D.; Hill, J.A. Can HFpEF and HFrEF coexist? Circulation 2020, 141, 709–711. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A. Cellular and molecular differences between HFpEF and HFrEF: A step ahead in an improved pathological understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Ruben, R.J. Unsolved issues around critical periods with emphasis on clinical application. Acta Oto-Laryngol. 1986, 101 (Suppl. 429), 61–64. [Google Scholar] [CrossRef]
- Murphy, S.P.; Ibrahim, N.E.; Januzzi, J.L. Heart failure with reduced ejection fraction: A review. Jama 2020, 324, 488–504. [Google Scholar] [CrossRef]
- Li, P.; Zhao, H.; Zhang, J.; Ning, Y.; Tu, Y.; Xu, D.; Zeng, Q. Similarities and differences between HFmrEF and HFpEF. Front. Cardiovasc. Med. 2021, 8, 678614. [Google Scholar] [CrossRef]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-inflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2018, 7, e007385. [Google Scholar] [CrossRef]
- DeBerge, M.; Shah, S.J.; Wilsbacher, L.; Thorp, E.B. Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol. Med. 2019, 25, 328–340. [Google Scholar] [CrossRef]
- Chen, G.; Bracamonte-Baran, W.; Diny, N.L.; Hou, X.; Talor, M.V.; Fu, K.; Liu, Y.; Davogustto, G.; Vasquez, H.; Taegtmeyer, H.; et al. Sca-1+ cardiac fibroblasts promote development of heart failure. Eur. J. Immunol. 2018, 48, 1522–1538. [Google Scholar] [CrossRef] [Green Version]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.; Chan, C.T.; Iwamoto, Y.; et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; Norris, P.C.; Kain, V.; Serhan, C.N.; Ingle, K.A. Splenic leukocytes define the resolution of inflammation in heart failure. Sci. Signal. 2018, 11, eaao1818. [Google Scholar] [CrossRef] [Green Version]
- Leuschner, F.; Rauch, P.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.; et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Deswal, A.; Petersen, N.; Feldman, A.; Young, J.; White, B.; Mann, D. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [Green Version]
- Testa, M.; Yeh, M.; Lee, P.; Berman, J.W.; Lejemtel, T.H.; Fanelli, R.; Loperfido, F. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol. 1996, 28, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Dunlay, S.M.; VRoger, L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Shrestha, K.; Sheehey, B.; Li, X.S.; Guggilam, A.; Wu, Y.; Finucan, M.; Gabi, A.; Medert, C.M.; Westfall, K.; et al. Elevated plasma marinobufagenin, an endogenous cardiotonic steroid, is associated with right ventricular dysfunction and nitrative stress in heart failure. Circ. Heart Fail. 2015, 8, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Haller, S.T.; Kennedy, D.J.; Shidyak, A.; Budny, G.V.; Malhotra, D.; Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am. J. Hypertens. 2012, 25, 690–696. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.J.; Elkareh, J.; Shidyak, A.; Shapiro, A.P.; Smaili, S.; Mutgi, K.; Gupta, S.; Tian, J.; Morgan, E.; Khouri, S.; et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am. J. Physiol.-Ren. Physiol. 2008, 294, F450–F454. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Models Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Frangogiannis, N.G. The Role of Macrophages in Nonischemic Heart Failure; American College of Cardiology Foundation: Washington, DC, USA, 2018; pp. 245–248. [Google Scholar]
- Shen, J.-l.; Xie, X.-j. Insight into the pro-inflammatory and profibrotic role of macrophage in heart failure with preserved ejection fraction. J. Cardiovasc. Pharmacol. 2020, 76, 276–285. [Google Scholar] [CrossRef]
- Liao, X.; Shen, Y.; Zhang, R.; Sugi, K.; Vasudevan, N.T.; Alaiti, M.A.; Sweet, D.R.; Zhou, L.; Qing, Y.; Gerson, S.L.; et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2018, 115, E4661–E4669. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Frangogiannis, N.G. Macrophages in the remodeling failing heart. Am. Heart Assoc. 2016, 119, 776–778. [Google Scholar] [CrossRef] [Green Version]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Fan, Q.; Tao, R.; Zhang, H.; Xie, H.; Lu, L.; Wang, T.; Su, M.; Hu, J.; Zhang, Q.; Chen, Q.; et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation 2019, 139, 663–678. [Google Scholar] [CrossRef]
- Liu, D.; Yan, W.; Huang, J.; Zhao, J.; Kilby, H.; Christopher, T.A.; Lopez, B.; Tao, L.; Ma, X.; Gu, G.; et al. Macrophages in ischemic heart failure: Yesterday, today, and tomorrow. Cardiol. Discov. 2021, 1, 128–134. [Google Scholar] [CrossRef]
- O’Rourke, S.A.; Dunne, A.; Monaghan, M.G. The role of macrophages in the infarcted myocardium: Orchestrators of ECM remodeling. Front. Cardiovasc. Med. 2019, 6, 101. [Google Scholar] [CrossRef] [Green Version]
- Di Corleto, P.E.; Chisolm, G., 3rd. Participation of the endothelium in the development of the atherosclerotic plaque. Prog. Lipid Res. 1986, 25, 365–374. [Google Scholar] [CrossRef]
- Chiu, J.-J.; Usami, S.; Chien, S. Vascular endothelial responses to altered shear stress: Pathologic implications for atherosclerosis. Ann. Med. 2009, 41, 19–28. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 2017, 97, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an inflammatory disease. Curr. Pharm. Des. 2012, 18, 4266–4288. [Google Scholar] [CrossRef]
- Skuratovskaia, D.A.; Vulf, M.; Komar, A.; Kirienkova, E.; Litvinova, L. Epigenetic regulation as a promising tool for treatment of atherosclerosis. Front. Biosci. 2020, 12, 173–199. [Google Scholar] [CrossRef]
- Herbin, O.; Regelmann, A.G.; Ramkhelawon, B.; Weinstein, E.G.; Moore, K.J.; Alexandropoulos, K. Monocyte adhesion and plaque recruitment during atherosclerosis development is regulated by the adapter protein Chat-H/SHEP1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1791–1801. [Google Scholar] [CrossRef] [Green Version]
- Pardali, E.; Makowski, L.M.; Leffers, M.; Borgscheiper, A.; Waltenberger, J. BMP-2 induces human mononuclear cell chemotaxis and adhesion and modulates monocyte-to-macrophage differentiation. J. Cell. Mol. Med. 2018, 22, 5429–5438. [Google Scholar] [CrossRef] [Green Version]
- Ray, M. Genetic Deletion of Interleukin-19 Exacerbates Atherogenesis in Double Knockout Mice by Modulation of mRNA Stability Protein HuR; Temple University: Setagaya, Tokyo, 2018. [Google Scholar]
- Vallet-Courbin, A.; Larivière, M.; Hocquellet, A.; Hémadou, A.; Parimala, S.-N.; Laroche-Traineau, J.; Santarelli, X.; Clofent-Sanchez, G.; Jacobin-Valat, M.-J.; Noubhani, A. A recombinant human anti-platelet SCFV antibody produced in pichia pastoris for atheroma targeting. PLoS ONE 2017, 12, e0170305. [Google Scholar] [CrossRef] [Green Version]
- Pirahanchi, Y.; Sinawe, H.; Dimri, M. Biochemistry, LDL Cholesterol; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef]
- Sallam, T.; Sandhu, J.; Tontonoz, P. Long noncoding RNA discovery in cardiovascular disease: Decoding form to function. Circ. Res. 2018, 122, 155–166. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, J.; Xu, X.; Qu, Y.; Dong, H.; Dang, J.; Huo, Z.; Xu, G. LncRNA expression profile during autophagy and Malat1 function in macrophages. PLoS ONE 2019, 14, e0221104. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, H.; Huang, S.; Yin, L.; Wang, F.; Luo, P.; Huang, H. Epigenetic regulation in cardiovascular disease: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2022, 7, 1–28. [Google Scholar] [CrossRef]
- Simion, V.; Haemmig, S.; Feinberg, M.W. LncRNAs in vascular biology and disease. Vasc. Pharmacol. 2019, 114, 145–156. [Google Scholar] [CrossRef]
- Simion, V.; Zhou, H.; Haemmig, S.; Pierce, J.B.; Mendes, S.; Tesmenitsky, Y.; Pérez-Cremades, D.; Lee, J.F.; Chen, A.F.; Ronda, N.; et al. A macrophage-specific lncRNA regulates apoptosis and atherosclerosis by tethering HuR in the nucleus. Nat. Commun. 2020, 11, 6135. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.-F.; Li, J.-L.; Peng, R.-J.; Liang, X.-Y.; Chen, X.-D.; Jiang, G.-H.; Shi, J.-F.; Si-Ma, Y.-H.; Xu, S.-Q. Mechanism of hyperproteinemia-induced blood cell homeostasis imbalance in an animal model. Zool Res. 2022, 18, 301–318. [Google Scholar] [CrossRef]
- Yu, X.H.; Deng, W.Y.; Chen, J.J.; Xu, X.D.; Liu, X.X.; Chen, L.; Shi, M.W.; Liu, Q.X.; Tao, M.; Ren, K. LncRNA kcnq1ot1 promotes lipid accumulation and accelerates atherosclerosis via functioning as a ceRNA through the miR-452-3p/HDAC3/ABCA1 axis. Cell Death Dis. 2020, 11, 1043. [Google Scholar] [CrossRef]
- Kojima, Y.; Volkmer, J.-P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J.; et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016, 536, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Zheng, L.; Qin, H.; Wang, W. Myocardial Infarction–associated Transcript Knockdown Inhibits Cell Proliferation, Migration, and Invasion Through miR-490-3p/Intercellular Adhesion Molecule 1 Axis in Oxidized Low-density Lipoprotein-induced Vascular Smooth Muscle Cells. J. Cardiovasc. Pharmacol. 2020, 76, 617–626. [Google Scholar] [CrossRef]
- Gao, Y.; Yue, J.; Huang, Z. LncRNA MIAT Mediates ox-LDL-Induced Endothelial Cell Injury Via miR-206/RAB22A Axis. J. Surg. Res. 2021, 265, 303–312. [Google Scholar] [CrossRef]
- Li, T.; Tu, P.; Bi, J.; Sun, Y.; Yu, D.; Wang, J.; Zhao, B. LncRNA Miat knockdown alleviates endothelial cell injury through regulation of miR-214-3p/Caspase-1 signalling during atherogenesis. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1231–1238. [Google Scholar] [CrossRef]
- Zhong, X.; Ma, X.; Zhang, L.; Li, Y.; Li, Y.; He, R. MIAT promotes proliferation and hinders apoptosis by modulating miR-181b/STAT3 axis in ox-LDL-induced atherosclerosis cell models. Biomed. Pharmacother. 2018, 97, 1078–1085. [Google Scholar] [CrossRef]
- Ye, Z.-M.; Yang, S.; Xia, Y.-P.; Hu, R.-T.; Chen, S.; Li, B.-W.; Chen, S.-L.; Luo, X.-Y.; Mao, L.; Li, Y.; et al. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 2019, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- Fasolo, F.; Jin, H.; Winski, G.; Chernogubova, E.; Pauli, J.; Winter, H.; Li, D.Y.; Glukha, N.; Bauer, S.; Metschl, S.; et al. Long noncoding RNA MIAT controls advanced Atherosclerotic lesion formation and plaque destabilization. Circulation 2021, 144, 1567–1583. [Google Scholar] [CrossRef]
- Jeries, H.; Volkova, N.; Grajeda-Iglesias, C.; Najjar, M.; Rosenblat, M.; Aviram, M.; Hayek, T. Prednisone and its active metabolite prednisolone attenuate lipid accumulation in macrophages. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 174–186. [Google Scholar] [CrossRef]
- Cohen, D.M.; Steger, D.J. Nuclear receptor function through genomics: Lessons from the glucocorticoid receptor. Trends Endocrinol. Metab. 2017, 28, 531–540. [Google Scholar] [CrossRef]
- Kim, J.-D.; Zhu, L.; Sun, Q.; Fang, L. Systemic metabolite profiling reveals sexual dimorphism of AIBP control of metabolism in mice. PLoS ONE 2021, 16, e0248964. [Google Scholar] [CrossRef]
- Bandaru, S.; Ala, C.; Salimi, R.; Akula, M.K.; Ekstrand, M.; Devarakonda, S.; Karlsson, J.; Eynden, J.V.D.; Bergström, G.; Larsson, E.; et al. Targeting filamin A reduces macrophage activity and atherosclerosis. Circulation 2019, 140, 67–79. [Google Scholar] [CrossRef]
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038. [Google Scholar] [CrossRef]
- Leung, R.; Wang, Y.; Cuddy, K.; Sun, C.; Magalhaes, J.; Grynpas, M.; Glogauer, M. Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J. Bone Miner. Res. 2010, 25, 1077–1091. [Google Scholar] [CrossRef]
- Khan, O.M.; Akula, M.K.; Skålen, K.; Karlsson, C.; Ståhlman, M.; Young, S.G.; Borén, J.; Bergo, M.O. Targeting GGTase-I activates RHOA, increases macrophage reverse cholesterol transport, and reduces atherosclerosis in mice. Circulation 2013, 127, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, G.; Solanki, S.; Dube, P.; Smedlund, K.; Ampem, P. On the roles of the transient receptor potential canonical 3 (TRPC3) channel in endothelium and macrophages: Implications in atherosclerosis. In Calcium Entry Pathways in Non-Excitable Cells; Springer: Berlin/Heidelberg, Germany, 2016; pp. 185–199. [Google Scholar]
- Dube, P.R.; Chikkamenahalli, L.L.; Birnbaumer, L.; Vazquez, G. Reduced calcification and osteogenic features in advanced atherosclerotic plaques of mice with macrophage-specific loss of TRPC3. Atherosclerosis 2018, 270, 199–204. [Google Scholar] [CrossRef]
- Solanki, S.; Dube, P.R.; Tano, J.-Y.; Birnbaumer, L.; Vazquez, G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am. J. Physiol.-Cell Physiol. 2014, 307, C521–C531. [Google Scholar] [CrossRef] [Green Version]
- Solanki, S.; Dube, P.R.; Birnbaumer, L.; Vazquez, G. Reduced necrosis and content of apoptotic M1 macrophages in advanced atherosclerotic plaques of mice with macrophage-specific loss of Trpc3. Sci. Rep. 2017, 7, 42526. [Google Scholar] [CrossRef]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Santovito, D.; Marcantonio, P.; Mastroiacovo, D.; Natarelli, L.; Mandolini, C.; De Nardis, V.; Paganelli, C.; De Cesare, D.; Affaitati, G.; Giamberardino, M.A.; et al. High dose rosuvastatin increases ABCA1 transporter in human atherosclerotic plaques in a cholesterol-independent fashion. Int. J. Cardiol. 2020, 299, 249–253. [Google Scholar] [CrossRef]
- Kuroda, K.; Otake, H.; Shinohara, M.; Kuroda, M.; Tsuda, S.; Toba, T.; Nagano, Y.; Toh, R.; Ishida, T.; Shinke, T.; et al. Effect of rosuvastatin and eicosapentaenoic acid on neoatherosclerosis: The LINK-IT Trial. EuroIntervention 2019, 15, e1099–e1106. [Google Scholar] [CrossRef] [Green Version]
- Sugizaki, Y.; Otake, H.; Kuroda, K.; Kawamori, H.; Toba, T.; Nagasawa, A.; Takeshige, R.; Nakano, S.; Matsuoka, Y.; Tanimura, K.; et al. Concomitant use of rosuvastatin and eicosapentaenoic acid significantly prevents native coronary atherosclerotic progression in patients with in-stent neoatherosclerosis. Circ. J. 2020, 84, 1826–1836. [Google Scholar] [CrossRef]
- Endo, J.; Sano, M.; Isobe, Y.; Fukuda, K.; Kang, J.X.; Arai, H.; Arita, M. 18-HEPE, an n-3 fatty acid metabolite released by macrophages, prevents pressure overload–induced maladaptive cardiac remodeling. J. Exp. Med. 2014, 211, 1673–1687. [Google Scholar] [CrossRef]
- Laguna-Fernandez, A.; Checa, A.; Carracedo, M.; Artiach, G.; Petri, M.H.; Baumgartner, R.; Forteza, M.J.; Jiang, X.; Andonova, T.; Walker, M.E.; et al. ERV1/ChemR23 signaling protects against atherosclerosis by modifying oxidized low-density lipoprotein uptake and phagocytosis in macrophages. Circulation 2018, 138, 1693–1705. [Google Scholar] [CrossRef]
- Geng, C.; Zhang, Y.; Hidru, T.H.; Zhi, L.; Tao, M.; Zou, L.; Chen, C.; Li, H.; Liu, Y. Sonodynamic therapy: A potential treatment for atherosclerosis. Life Sci. 2018, 207, 304–313. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, T.; Sun, X.; Liu, M.; Shen, Z.; Li, B.; Zhao, X.; Jin, H.; Zhang, Z.; Tian, Y. Membrane-permeabilized sonodynamic therapy enhances drug delivery into macrophages. PLoS ONE 2019, 14, e0217511. [Google Scholar] [CrossRef]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Kaikita, K.; Hayasaki, T.; Okuma, T.; Kuziel, W.A.; Ogawa, H.; Takeya, M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 2004, 165, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Xu, A.; Sun, X.; Yang, Y.; Zhang, L.; Bai, H.; Ben, J.; Zhu, X.; Li, X.; Wang, Z.; et al. Self-maintenance of cardiac resident reparative macrophages attenuates doxorubicin-induced cardiomyopathy through the SR-A1-C-MYC axis. Circ. Res. 2020, 127, 610–627. [Google Scholar] [CrossRef]
- Xu, M.; Li, X.; Song, L. Baicalin regulates macrophages polarization and alleviates myocardial ischaemia/reperfusion injury via inhibiting JAK/STAT pathway. Pharm. Biol. 2020, 58, 655–663. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Hu, J.; Chen, F.; Qiao, S.; Sun, X.; Gao, L.; Xie, J.; Xu, B. Mesenchymal stromal cell-derived exosomes attenuate myocardial ischaemia-reperfusion injury through miR-182-regulated macrophage polarization. Cardiovasc. Res. 2019, 115, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Zhang, S.; Tang, T.-T.; Cheng, X. Bioinformatics and Immune Infiltration Analyses Reveal the Key Pathway and Immune Cells in the Pathogenesis of Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 696321. [Google Scholar] [CrossRef]
- Schultheiss, H.-P.; Baumeier, C.; Pietsch, H.; Bock, C.-T.; Poller, W.; Escher, F. Cardiovascular consequences of viral infections: From COVID to other viral diseases. Cardiovasc. Res. 2021, 117, 2610–2623. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mohiddin, S.A.; DiMarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef]
- Xue, Y.L.; Zhang, S.X.; Zheng, C.F.; Li, Y.F.; Zhang, L.H.; Su, Q.Y.; Hao, Y.F.; Wang, S.; Li, X.W. Long non-coding RNA MEG3 inhibits M2 macrophage polarization by activating TRAF6 via microRNA-223 down-regulation in viral myocarditis. J. Cell. Mol. Med. 2020, 24, 12341–12354. [Google Scholar] [CrossRef]
- Wu, X.; Meng, Y.; Wang, C.; Yue, Y.; Dong, C.; Xiong, S. Semaphorin7A aggravates coxsackievirusB3-induced viral myocarditis by increasing α1β1-integrin macrophages and subsequent enhanced inflammatory response. J. Mol. Cell. Cardiol. 2018, 114, 48–57. [Google Scholar] [CrossRef]
- Sanchez-Jimenez, E.F. Initial clinical presentation of Takotsubo cardiomyopathy with-a focus on electrocardiographic changes: A literature review of cases. World J. Cardiol. 2013, 5, 228. [Google Scholar] [CrossRef]
- Scally, C.; Abbas, H.; Ahearn, T.; Srinivasan, J.; Mezincescu, A.; Rudd, A.; Spath, N.; Yucel-Finn, A.; Yuecel, R.; Oldroyd, K.; et al. Myocardial and systemic inflammation in acute stress-induced (Takotsubo) cardiomyopathy. Circulation 2019, 139, 1581–1592. [Google Scholar] [CrossRef]
- Nishida, K.; Otsu, K. Inflammation and metabolic cardiomyopathy. Cardiovasc. Res. 2017, 113, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Yan, D.; Yang, L.; Sun, Y.; Zhan, L.; Lu, L.; Jin, Z.; Zhang, C.; Long, P.; Chen, J.; et al. The effect of miR-471-3p on macrophage polarization in the development of diabetic cardiomyopathy. Life Sci. 2021, 268, 118989. [Google Scholar] [CrossRef]
- Wiese, S.; Voiosu, A.; Hove, J.D.; Danielsen, K.V.; Voiosu, T.; Grønbaek, H.; Møller, H.J.; Genovese, F.; Reese-Petersen, A.L.; Mookerjee, R.P.; et al. Fibrogenesis and inflammation contribute to the pathogenesis of cirrhotic cardiomyopathy. Aliment. Pharmacol. Ther. 2020, 52, 340–350. [Google Scholar] [CrossRef]
- Li, D.; Yang, Y.; Wang, S.; He, X.; Liu, M.; Bai, B.; Tian, C.; Sun, R.; Yu, T.; Chu, X. Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol. 2021, 46, 102089. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Hanna, A.; Frangogiannis, N.G. Inflammatory cytokines and chemokines as therapeutic targets in heart failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef]
- Ceneri, N.; Zhao, L.; Young, B.D.; Healy, A.; Coskun, S.; Vasavada, H.; Yarovinsky, T.O.; Ike, K.; Pardi, R.; Qin, L.; et al. Rac2 modulates atherosclerotic calcification by regulating macrophage interleukin-1β production. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Van Tassell, B.W.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Oddi Erdle, C.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 blockade in recently decompensated systolic heart failure: Results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e004373. [Google Scholar] [CrossRef]
- Buckley, L.F.; Carbone, S.; Trankle, C.R.; Canada, J.M.; Erdle, C.O.; Regan, J.A.; Viscusi, M.M.; Kadariya, D.; Billingsley, H.; Arena, R.; et al. Effect of Interleukin-1 Blockade on Left Ventricular Systolic Performance and Work: A Post-Hoc Pooled Analysis of Two Clinical Trials. J. Cardiovasc. Pharmacol. 2018, 72, 68. [Google Scholar] [CrossRef]
- Yokoe, I.; Kobayashi, H.; Giles, J.; Yoneyama, K.; Kitamura, N.; Takei, M. Impact of tocilizumab on N-terminal pro-brain natriuretic peptide levels in patients with active rheumatoid arthritis without cardiac symptoms. Scand. J. Rheumatol. 2018, 47, 364–370. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Zheng, Y.; Qin, L.; Zacarías, N.V.O.; De Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Goody, P.R.; Hosen, M.R.; Christmann, D.; Niepmann, S.T.; Zietzer, A.; Adam, M.; Bönner, F.; Zimmer, S.; Nickenig, G.; Jansen, F. Aortic valve stenosis: From basic mechanisms to novel therapeutic targets. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 885–900. [Google Scholar] [CrossRef]
- Johnson, T.A.; Singla, D.K. PTEN inhibitor VO-OHpic attenuates inflammatory M1 macrophages and cardiac remodeling in doxorubicin-induced cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2018, 315, H1236–H1249. [Google Scholar] [CrossRef]
- Munjal, C.; Jegga, A.; Opoka, A.M.; Stoilov, I.; Norris, R.A.; Thomas, C.J.; Smith, J.M.; Mecham, R.P.; Bressan, G.M.; Hinton, R.B. Inhibition of MAPK-Erk pathway in vivo attenuates aortic valve disease processes in Emilin1-deficient mouse model. Physiol. Rep. 2017, 5, e13152. [Google Scholar] [CrossRef]
- Zhao, M.; Li, F.; Jian, Y.; Wang, X.; Yang, H.; Wang, J.; Su, J.; Lu, X.; Xi, M.; Wen, A.; et al. Salvianolic acid B regulates macrophage polarization in ischemic/reperfused hearts by inhibiting mTORC1-induced glycolysis. Eur. J. Pharmacol. 2020, 871, 172916. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Xu, N.; Umeyama, K.; Hulin, A.; Ponny, S.R.; Vagnozzi, R.J.; Green, E.A.; Hanson, P.; McManus, B.M.; Nagashima, H.; et al. Deficiency of circulating monocytes ameliorates the progression of myxomatous valve degeneration in Marfan syndrome. Circulation 2020, 141, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Kim, E.-Y.; Kim, J.-E.; Oh, S.; Park, S.-O.; Kim, S.-M.; Choi, H.; Song, J.-K.; Chang, E.-J. Evogliptin suppresses calcific aortic valve disease by attenuating inflammation, fibrosis, and calcification. Cells 2021, 10, 57. [Google Scholar] [CrossRef]
- Nguyen, P.A.; Won, J.S.; Rahman, M.K.; Bae, E.J.; Cho, M.K. Modulation of Sirt1/NF-κB interaction of evogliptin is attributed to inhibition of vascular inflammatory response leading to attenuation of atherosclerotic plaque formation. Biochem. Pharmacol. 2019, 168, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Kain, D.; Amit, U.; Yagil, C.; Landa, N.; Naftali-Shani, N.; Molotski, N.; Aviv, V.; Feinberg, M.S.; Goitein, O.; Kushnir, T.; et al. Macrophages dictate the progression and manifestation of hypertensive heart disease. Int. J. Cardiol. 2016, 203, 381–395. [Google Scholar] [CrossRef]
- Gallet, R.; Dawkins, J.; Valle, J.; Simsolo, E.; De Couto, G.; Middleton, R.; Tseliou, E.; Luthringer, D.; Kreke, M.; Smith, R.R.; et al. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur. Heart J. 2017, 38, 201–211. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, T.W.; Khalaf, F.K.; Soehnlen, S.; Hegde, P.; Storm, K.; Meenakshisundaram, C.; Dworkin, L.D.; Malhotra, D.; Haller, S.T.; Kennedy, D.J.; et al. Dirty Jobs: Macrophages at the Heart of Cardiovascular Disease. Biomedicines 2022, 10, 1579. https://doi.org/10.3390/biomedicines10071579
Stevens TW, Khalaf FK, Soehnlen S, Hegde P, Storm K, Meenakshisundaram C, Dworkin LD, Malhotra D, Haller ST, Kennedy DJ, et al. Dirty Jobs: Macrophages at the Heart of Cardiovascular Disease. Biomedicines. 2022; 10(7):1579. https://doi.org/10.3390/biomedicines10071579
Chicago/Turabian StyleStevens, Travis W., Fatimah K. Khalaf, Sophia Soehnlen, Prajwal Hegde, Kyle Storm, Chandramohan Meenakshisundaram, Lance D. Dworkin, Deepak Malhotra, Steven T. Haller, David J. Kennedy, and et al. 2022. "Dirty Jobs: Macrophages at the Heart of Cardiovascular Disease" Biomedicines 10, no. 7: 1579. https://doi.org/10.3390/biomedicines10071579
APA StyleStevens, T. W., Khalaf, F. K., Soehnlen, S., Hegde, P., Storm, K., Meenakshisundaram, C., Dworkin, L. D., Malhotra, D., Haller, S. T., Kennedy, D. J., & Dube, P. (2022). Dirty Jobs: Macrophages at the Heart of Cardiovascular Disease. Biomedicines, 10(7), 1579. https://doi.org/10.3390/biomedicines10071579