
Drug Repurposing: The Mechanisms and Signaling Pathways of Anti-Cancer Effects of Anesthetics


Abstract
:1. Introduction
2. Repurposing Anesthetic/Sedative Drugs into Anticancer Treatment
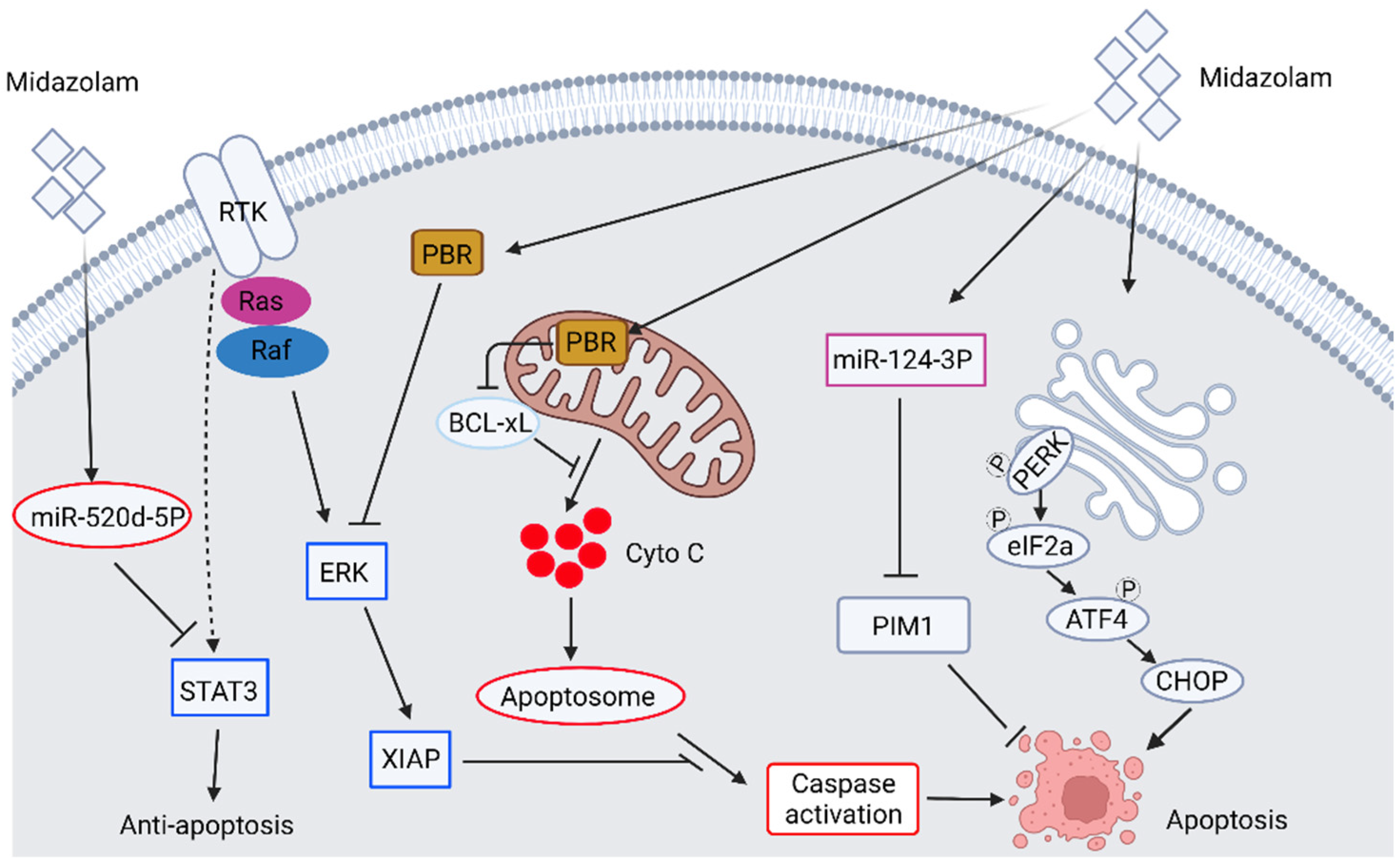
2.1. The Anticancer Ability of Benzodiazepine Derivative Midazolam
2.2. The Potential Antipsychotic Agents in Anticancer Studies: Role of Haloperidol and Its Derivatives
2.3. The Anti-Cancer Ability of Ketamine
2.4. The Application of Lidocaine and Its Similar Drugs in the Anti-Cancer Study
2.5. The Application of Propofol in Cancer Treatment
2.6. The Effect of Valproic Acid in Anti-Cancer Therapy
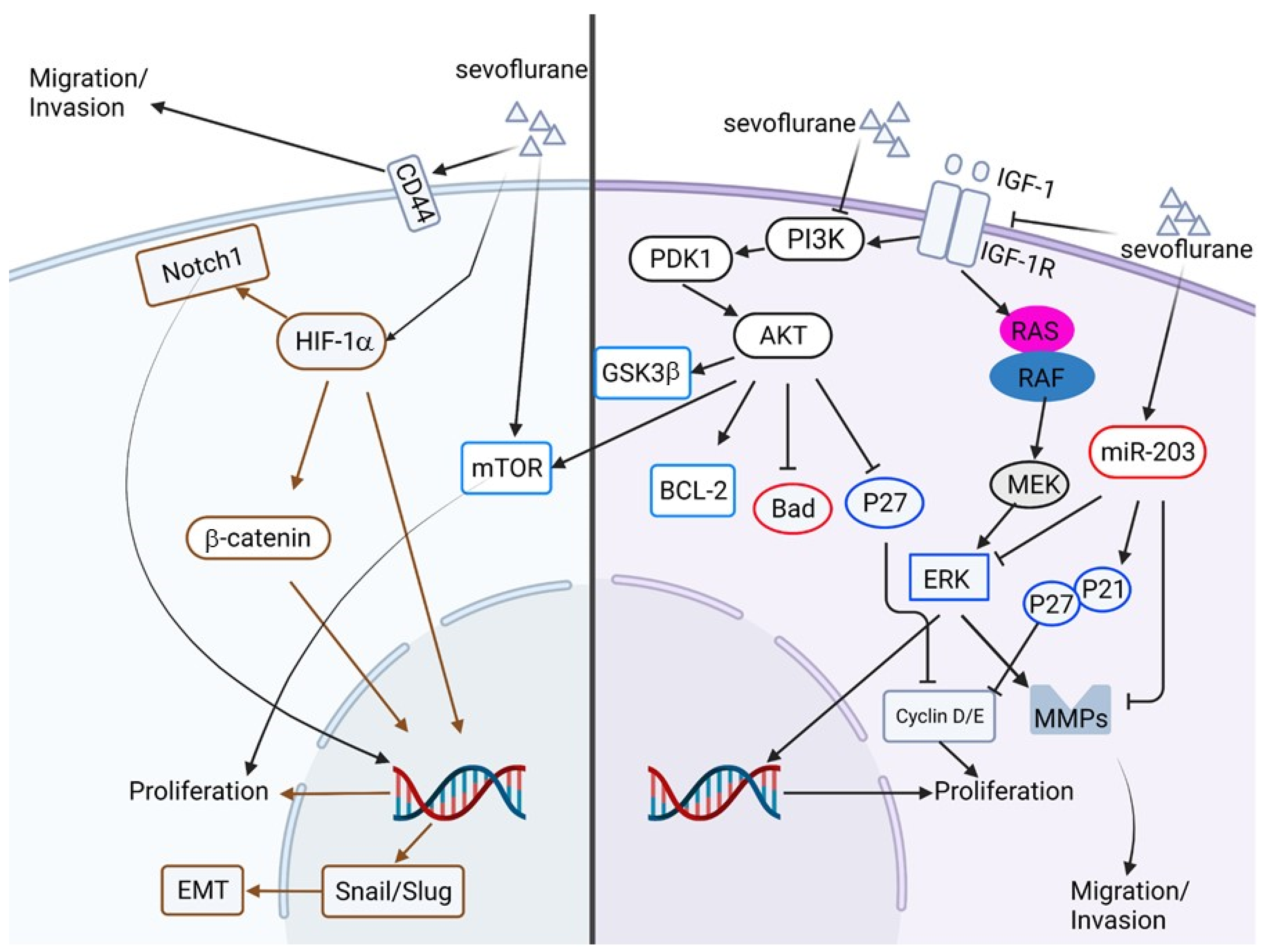
2.7. The Anti-Cancer Potential of Volatile Anesthetics
3. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.B.; Anderson, R.N. The leading causes of death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Li, S.D. Modifying the tumor microenvironment using nanoparticle therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 891–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GlobalSurg Collaborative and National Institute for Health Research Global Health Research Unit on Global Surgery. Global variation in postoperative mortality and complication after cancer surgery: A multicenter, prospective cohort study in 82 countries. Lancet 2021, 397, 387–397. [Google Scholar] [CrossRef]
- Gatta, G.; Zigon, G.; Capocaccia, R.; Coebergh, J.W.; Desandes, E.; Kaatsch, P.; Pastore, G.; Peris-Bonet, R.; Stiller, C.A. Survival of European children and young adults with cancer diagnosed 1995–2002. Eur. J. Cancer 2009, 45, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Mailankody, S. Research and development spending to bring a single cancer drug to market and revenues after approval. JAMA Intern. Med. 2017, 177, 1569–1575. [Google Scholar] [CrossRef]
- Dusetzina, S.B. Drug pricing trends for orally administered anticancer medications reimbursed by commercial health plans, 2000–2014. JAMA Oncol. 2016, 2, 960–961. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Tim Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.S.; Carpistrano, I.R.; Crispino, S.; Gyawali, B.; Rooman, I.; Nuffel, A.M.V.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The repurposing drugs in oncology database. Ecancer 2018, 12, 886. [Google Scholar] [CrossRef] [Green Version]
- Pantziarka, P.; Vandeborne, L.; Bouche, G. A database of drug repurposing clinical trials in oncology. Front. Pharmacol. 2021, 12, 790952. [Google Scholar] [CrossRef]
- Wall, T.; Sherwin, A.; Ma, D.; Buggy, J. Influence of perioperative anaesthetic and analgesic interventions on oncological outcomes: A narrative review. Br. J. Anesth. 2019, 123, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Olkkola, K.T.; Ahonen, J. Midazolam and other benzodiazepines. Handb. Exp. Pharmacol. 2008, 182, 335–360. [Google Scholar]
- Möhler, H.; Fritschy, J.M.; Rudolp, U. A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 2002, 300, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieghart, W. Structure and pharmacology of gamma-aminobutyric acid receptor subtypes. Pharmacol. Rev. 1995, 47, 181–234. [Google Scholar] [PubMed]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M., Jr.; Kim, J.J.; Hibbs, R.E. Structure of a human synaptic GABAA receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef]
- Lacapere, J.J.; Papadopoulos, V. Peripheral-type benzodiazepine receptor: Structure and function of a cholesterol binding protein in steroid and bile acid biosynthesis. Steroids 2003, 68, 569–585. [Google Scholar] [CrossRef]
- Kumar, A.; Muzik, O.; Chugani, D.; Chakraborty, P.; Chugani, H.T. PET-derived biodistribution and dosimetry of the benzodiazepine receptor-bionding radioligand 11C-(R)-PK11195 in children and adults. J. Nucl. Med. 2010, 51, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, M.; Fertikh, D.; Culty, M.; Li, H.; Vidic, B.; Papadopoulos, V. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: Correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999, 59, 831–842. [Google Scholar]
- Olson, J.M.; Ciliax, B.J.; Mancini, W.R.; Young, A.B. Presence of peripheral-type benzodiazepine binding sites on human erythrocyte membranes. Eur. J. Pharmacol. 1988, 152, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Oke, B.O.; Suarez-Quian, C.A.; Riond, J.; Ferrara, P.; Papadopoulos, V. Cell surface localization of the peripheral-type benzodiazepine receptor (PBR) in adrenal cortex. Mol. Cell. Endocrinol. 1992, 87, R1–R6. [Google Scholar] [CrossRef]
- Casellas, P.; Galiegue, S.; Basile, A.S. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Intl. 2002, 40, 475–486. [Google Scholar] [CrossRef]
- Corsi, L.; Geminiani, E.; Avallone, R.; Baraldi, M. Nuclear location-dependent role of peripheral benzodiazepine receptor (PBR) in hepatic tumoral cell lines proliferation. Life Sci. 2005, 76, 2523–2533. [Google Scholar] [CrossRef] [PubMed]
- Beinlich, A.; Strohmeier, R.; Kaufmann, M.; Kuhl, H. Relation of cell proliferation to expression of peripheral benzodiazepine receptors in human breast cancer cell lines. Biochem. Pharmacol. 2000, 60, 397–402. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Amri, H.; Boujrad, N.; Cascio, C.; Culty, M.; Gamier, M.; Hardwick, M.; Li, H.; Vidic, B.; Brown, A.S.; et al. Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 1997, 62, 21–28. [Google Scholar] [CrossRef]
- Culty, M.; Li, H.; Boujrad, N.; Amri, H.; Vidic, B.; Bernassau, J.M.; Reversat, J.L.; Papadopoulos, V. In vitro studies on the role of the peripheral-type benzodiazepine receptor in steroidogenesis. J. Steroid Biochem. Mol. Biol. 1999, 69, 123–130. [Google Scholar] [CrossRef]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef]
- Azarashvili, T.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Yurkov, I.; Papadopoulos, V.; Reiser, G. The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeability transition pore opening in rat brain mitochondria. Cell Calcium 2007, 42, 27–39. [Google Scholar] [CrossRef]
- Lee, J.J.; Eisenberg, P.; Papadopoulos, V.; Wang, J.; Widmaier, E.P. Reversible changes in adrenocorticotropin (ACTH)-induced adrenocortical steroidogenesis and expression of the peripheral-type benzodiazepine receptor during the ACTH-insensitive period in young rats. Endocrinology 2004, 145, 2165–2173. [Google Scholar] [CrossRef] [Green Version]
- Sutter, A.P.; Maaser, K.; Gerst, B.; Krahn, A.; Zeitz, M.; Scherübl, H. Enhancement of peripheral benzodiazepine receptor ligand-induced apoptosis and cell cycle arrest of esophageal cancer cells by simultaneous inhibition of MAPK/ERK kinase. Biochem. Pharmacol. 2004, 67, 1701–1710. [Google Scholar] [CrossRef]
- Fischer, R.; Schmitt, M.; Bode, J.G.; Häussinger, D. Expression of the peripheral-type benzodiazepine receptor and apoptosis induction in hepatic stellate cells. Gastroenterology 2001, 120, 1212–1226. [Google Scholar] [CrossRef]
- So, E.C.; Lin, Y.X.; Tseng, C.H.; Pan, B.S.; Cheng, K.S.; Wong, K.L.; Hao, L.J.; Wang, Y.K.; Huang, B.M. Midazolam induces apoptosis in MA-10 mouse Leydig tumor cells through caspase activation and the involvement of MAPK signaling pathway. OncoTargets Ther. 2014, 7, 211–221. [Google Scholar] [PubMed] [Green Version]
- So, E.C.; Chen, Y.C.; Wang, S.C.; Wu, C.C.; Huang, M.C.; Lai, M.S.; Pan, B.S.; Kang, F.C.; Huang, B.M. Midazolam regulated caspase pathway, endoplasmic reticulum stress, autophagy, and cell cycle to induce apoptosis in MA-10 mouse Leydig tumor cells. OncoTargets Ther. 2016, 9, 2519–2533. [Google Scholar] [PubMed] [Green Version]
- Jiao, J.; Wang, Y.; Sun, X.; Jiang, X. Midazolam induces A549 cell apoptosis in vitro via the miR-520d-5p/STAT3 pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 1365–1373. [Google Scholar] [PubMed]
- Mishra, S.K.; Kang, J.H.; Lee, C.W.; Oh, S.H.; Ryu, J.S.; Bae, Y.S.; Kim, H.M. Midazolam induces cellular apoptosis in human cancer cells and inhibits tumor growth in xenograft mice. Mol. Cells 2013, 36, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Yao, X.; Du, X. Midazolam inhibits proliferation and accelerates apoptosis of hepatocellular carcinoma cells by elevating microRNA-124-3p and suppressing PIM-1. IUBMB Life 2020, 72, 452–464. [Google Scholar] [CrossRef]
- Stevens, M.F.; Werdehausen, R.; Gaza, N.; Hermanns, H.; Kremer, D.; Bauer, I.; Küry, P.; Hollmann, M.W.; Braun, S. Midazolam activates the intrinsic pathway of apoptosis independent of benzodiazepine and death receptor signaling. Reg. Anesth. Pain Med. 2011, 36, 343–349. [Google Scholar] [CrossRef]
- Sanomachi, T.; Suzuki, S.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Togashi, K.; Seino, S.; Yoshioka, T.; Okada, M.; Kitanaka, C. Olanzapine, an atypical antipsychotic, inhibits survivin expression and sensitizes cancer cells to chemotherapeutic agents. Anticancer Res. 2017, 37, 6177–6188. [Google Scholar]
- Xu, W.; Hu, J.; Liu, W.; Zhu, Q.; Gong, X.; Zhu, P.; Yang, X.; Xia, R.; Xue, R. Remimazolan inhibits glioma cell growth and induces apoptosis through down-regulation of NF-κB pathway. IUBMB Life 2021, 73, 341–348. [Google Scholar] [CrossRef]
- Wang, C.; Datoo, T.; Zhao, H.; Wu, L.; Date, A.; Jiang, C.; Sanders, R.D.; Wang, G.; Bevan, C.; Ma, D. Midazolam and dexmedetomidine affect neuroglioma and lung carcinoma cell biology in vitro and in vivo. Anesthesiology 2018, 129, 1000–1014. [Google Scholar] [CrossRef]
- Lu, H.L.; Wu, K.C.; Chen, C.W.; Weng, H.K.; Huang, B.M.; Lin, T.Y.; Liu, M.H.; So, E.C.; Lin, R.M.; Wang, Y.K. Anticancer Effects of Midazolam on Lung and Breast Cancers by Inhibiting Cell Proliferation and Epithelial-Mesenchymal Transition. Life 2021, 11, 1396. [Google Scholar] [CrossRef]
- Seo, J.A.; Jeon, H.Y.; Kim, M.; Lee, Y.J.; Han, E.T.; Park, W.S.; Hong, S.H.; Kim, Y.M.; Ha, K.S. Anti-metastatic effect of midazolam on melanoma B16F10 cells in the lungs of diabetic mice. Biochem. Pharmacol. 2020, 178, 114052. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Chen, J.; Sun, X.; Wang, G. Midazolam increases cisplatin-sensitivity in non-small cell lung cancer (NSCLC) via the miR-194-5p/HOOK3 axis. Cancer Cell Int. 2021, 21, 401. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Frank, C.L.; de Anda, F.C.; Tsai, L.H. Hook3 interacts with PCM1 to regulate pericentriolar material assembly and the timing of neurogenesis. Neuron 2010, 65, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villari, G.; Enrico Bena, C.; Del Giudice, M.; Gioelli, N.; Sandri, C.; Camillo, C.; Fiorio Pla, A.; Bosia, C.; Serini, G. Distinct retrograde microtubule motor sets drive early and late endosome transport. EMBO J. 2020, 39, e103661. [Google Scholar] [CrossRef] [PubMed]
- Oshima, Y.; Sano, M.; Kajiwara, I.; Ichimaru, Y.; Itaya, T.; Kuramochi, T.; Hayashi, E.; Kim, J.; Kitajima, O.; Masugi, Y.; et al. Midazolam exhibits antitumour and anti-inflammatory effects in a mouse model of pancreatic ductal adenocarcinoma. Br. J. Anesth. 2022, 128, 679–690. [Google Scholar] [CrossRef]
- Tyler, M.W.; Zaldivar-Diez, J.; Haggarty, S.J. Classics in Chemical Neuroscience: Haloperidol. ACS Chem. Neurosci. 2017, 15, 444–453. [Google Scholar] [CrossRef]
- Calver, L.; Drinkwater, V.; Gupta, R.; Page, C.B.; Isbister, G.K. Droperidol v. haloperidol for sedation of aggressive behaviour in acute mental health: Randomised controlled trial. Br. J. Psychiatry 2015, 206, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Gassó, P.; Mas, S.; Molina, O.; Bernardo, M.; Lafuente, A.; Parellada, E. Neurotoxic/neuroprotective activity of haloperidol, risperidone and paliperidone in neuroblastoma cells. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 36, 71–77. [Google Scholar] [CrossRef]
- Faraz, F.; Pannullo, S.; Rosenblum, M.; Smith, A.; Wernicke, A.G. Long-term survival in a patient with glioblastoma on antipsychotic therapy for schizophrenia: A case report and literature review. Ther. Adv. Med. Oncol. 2016, 8, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Baid, U.; Rane, S.U.; Taibal, S.; Gupta, S.; Thakur, M.H.; Moiyadi, A.; Mahajan, A. Overall survival prediction in glioblastoma with radiomic features using machine learning. Front. Comput. Neurosci. 2020, 14, 61. [Google Scholar] [CrossRef]
- Papadopoulos, F.; Isihou, R.; Alexiou, G.A.; Tsalios, T.; Vartholomatos, E.; Markopoulos, G.S.; Sioka, C.; Tsekeris, P.; Kyritsis, A.P.; Galani, V. Haloperidol induced cell cycle arrest and apoptosis in glioblastoma cells. Biomedicines 2020, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Qi, J.; Dai, Y.; Bowen, W.D.; Mousseau, D.D. Haloperidol disrupts Akt signalling to reveal a phosphorylation-dependent regulation of pro-apoptotic Bcl-XS function. Cell Signal. 2009, 21, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Asong, G.M.; Amissah, F.; Voshavar, C.; Nkembo, A.T.; Ntantie, E.; Lamango, N.S.; Ablordeppey, S.Y. A mechanistic investigation on the anticancer properties of sya013, a homopiperazine analogue of haloperidol with activity against triple negative breast cancer cells. ACS Omega 2020, 5, 32907–32918. [Google Scholar] [CrossRef]
- Kataoka, Y.; Ishikawa, M.; Miura, M.; Takeshita, M.; Fujita, R.; Furusawa, S.; Takayanagi, M.; Takayanagi, Y.; Sasaki, K. Reversal of vinblastine resistance in human leukemic cells by haloperidol and dihydrohaloperidol. Biol. Pharm. Bull. 2001, 24, 612–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, J. Ketamine in pain management. CNS Neurosci. Ther. 2013, 19, 396–402. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, M.; Wang, X.; Yang, X.; Wang, M.; Zhang, C.; Zhou, S.; Tang, N. Impact of ketamine on learning and memory function, neuronal apoptosis and its potential association with miR-214 and PTEN in adolescent rats. PLoS ONE 2014, 9, e99855. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Huang, Y.; Lu, Y.; Chen, J.; Jiang, H. Repeated administration of ketamine can induce hippocampal neurodegeneration and long-term cognitive impairment via the ROS/HIF-1α pathway in developing rats. Cell Physiol. Biochem. 2014, 33, 1715–1732. [Google Scholar] [CrossRef]
- Jimi, N.; Segawa, K.; Minami, K.; Sata, T.; Shigematsu, A. Inhibitory effect of the intravenous anesthetic, ketamine, on rat mesangial cell proliferation. Anesth. Analg. 1997, 84, 190–195. [Google Scholar] [CrossRef]
- Shiga, Y.; Minami, K.; Segawa, K.; Uezono, Y.; Shiraishi, M.; Sata, T.; Yamamoto, C.; Kim, S.T. The inhibition of aortic smooth muscle cell proliferation by the intravenous anesthetic ketamine. Anesth. Analg. 2004, 99, 1408–1412. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, P.; Luo, W.; Zhang, L.; Hu, R.; Sun, Y.; Jiang, H. Ketamine induces apoptosis in lung adenocarcinoma cells by regulating the expression of CD69. Cancer Med. 2018, 7, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, R.; Carracedo, J.; Castedo, M.; Zamzami, N.; Kroemer, G. CD69-induced monocyte apoptosis involves multiple nonredundant signaling pathways. Cell Immunol. 1996, 172, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, J.; Yang, B.; Zhao, G.; Lin, H.; Liu, Q.; Wang, L.; Wan, Y.; Jiang, H. Ketamine inhibits ovarian cancer cell growth by regulating the lncRNA-PVT1/EZH2/p57 Axis. Front. Genet. 2021, 11, 597467. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, W.; Zhang, X.; Wu, F.; Sun, D.; Wang, Z. Ketamine suppresses proliferation and induces ferroptosis and apoptosis of breast cancer cells by targeting KAT5/GPX4 axis. Biochem. Biophys. Res. Commun. 2021, 585, 111–116. [Google Scholar] [CrossRef]
- He, G.N.; Bao, N.R.; Wang, S.; Xi, M.; Zhang, T.H.; Chen, F.S. Ketamine induces ferroptosis of liver cancer cells by targeting lncRNA PVT1/miR-214-3p/GPX4. Drug Des. Devel. Ther. 2021, 15, 3965–3978. [Google Scholar] [CrossRef]
- Cho, J.S.; Kim, N.Y.; Shim, J.K.; Jun, J.H.; Lee, S.; Kwak, Y.L. The immunomodulatory effect of ketamine in colorectal cancer surgery: A randomized-controlled trial. Can. J. Anesth. 2021, 68, 683–692. [Google Scholar] [CrossRef]
- Kubota, M.; Niwa, H.; Seya, K.; Kawaguchi, J.; Kushikata, T.; Hirota, K. Ketamine does not change natural killer cell cytotoxicity in patients undergoing cancer surgery: Basic experiment and clinical trial. J. Oncol. 2022, 2022, 8946269. [Google Scholar] [CrossRef]
- Xi, Y.; Wei, X.; Mu, Y.; Li, Q.; Liu, J. A review of the mechanism of central analgesic effect of lidocaine. Medicine 2020, 99, e19898. [Google Scholar]
- Nouette-Gaulain, K.; Dadure, C.; Morau, D.; Pertuiset, C.; Galbes, O.; Hayot, M.; Mercier, J.; Sztark, F.; Rossignol, R.; Capdevila, X. Age-dependent bupivacaine-induced muscle toxicity during continuous peripheral nerve block in rats. Anesthesiology 2009, 111, 1120–1127. [Google Scholar] [CrossRef] [Green Version]
- Nouette-Gaulain, K.; Bellance, N.; Prevost, B.; Passerieux, E.; Pertuiset, C.; Galbes, O.; Smolkova, K.; Masson, F.; Miraux, S.; Delage, J.P.; et al. Erythropoietin protects against local anesthetic myotoxicity during continuous regional analgesia. Anesthesiology 2010, 110, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Xuan, W.; Zhao, H.; Hankin, J.; Chen, L.; Yao, S.; Ma, D. Local anesthetic bupivacaine induced ovarian and prostate cancer apoptotic cell death and underlying mechanisms in vitro. Sci. Rep. 2016, 6, 26277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Huang, S.; Gao, Y. lidocaine inhibits proliferation and metastasis of epithelial ovarian cancer through the Wnt/β-catenin pathway. Transl. Cancer Res. 2021, 10, 3479–3490. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Chen, D.T.; Pan, J.H.; Chen, Y.H.; Yan, Y.; Li, Q.; Xue, R.F.; Yuan, Y.F.; Zeng, W.A. Lidocaine induces apoptosis and suppresses tumor growth in human hepatocellular carcinoma cells in vitro and in a xenograft model in vitro. Anesthesiology 2017, 126, 868–881. [Google Scholar]
- Jose, C.; Hebert-Chatelain, E.; Dias, A.N.; Roche, E.; Obre, E.; Lacombe, D.; Rezvani, H.R.; Pourquier, P.; Nouette-Gaulain, K.; Rossignol, R. Redox mechanism of levobupivacaine cytostatic effect on human prostate cancer cells. Redox Biol. 2018, 18, 33–42. [Google Scholar] [CrossRef]
- Meng, M.; Huang, M.; Liu, C.; Wang, J.; Ren, W.; Cui, S.; Gu, J.; Xie, J.; Ma, B.; Yang, G.; et al. Local anesthetic levobupivacaine induces ferroptosis and inhibits progression by up-regulating p53 in non-small cell lung cancer. Aging 2021, 27, 13. [Google Scholar] [CrossRef]
- Mao, S.H.; Zhu, C.H.; Nie, Y.; Yu, J.; Wang, L. Levobupivacaine Induces Ferroptosis by miR-489-3p/SLC7A11 Signaling in Gastric Cancer. Front. Pharmacol. 2021, 12, 681338. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Fraga, M.F.; Espada, J.; Esteller, M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003, 63, 4984–4989. [Google Scholar]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Ramirez, M.F.; Tran, P.; Cata, J.P. The effect of clinically therapeutic plasma concentrations of lidocaine on natural killer cell cytotoxicity. Reg. Anesth. Pain Med. 2015, 40, 43–48. [Google Scholar] [CrossRef]
- Cata, J.P.; Ramirez, M.F.; Velasquez, J.F.; Di, A.I.; Popat, K.U.; Gottumukkala, V.; Black, D.M.; Lewis, V.O.; Vauthey, J.N. Lidocaine Stimulates the Function of Natural Killer Cells in Different Experimental Settings. Anticancer Res. 2017, 37, 4727–4732. [Google Scholar]
- Wei, Q.; Xia, M.; Zhang, Q.; Wang, Z. Effect of intravenous lidocaine infusion on perioperative cellular immunity and the quality of postoperative recovery in breast cancer patients: A randomized controlled trial. Gland Surg. 2022, 11, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Chidambaran, V.; Costandi, A.; D’Mello, A. Propofol: A review of its role in pediatric anesthesia and sedation. CNS Drugs 2015, 29, 543–563. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, Y.; Huang, L.; Zhang, F.; Kang, R. Effects of propofol on cancer development and chemotherapy: Potential mechanisms. Eur. J. Pharmacol. 2018, 831, 46–51. [Google Scholar] [CrossRef]
- Pirttikangas, C.O.; Salo, M.; Peltola, O. Propofol infusion anaesthesia and the immune response in elderly patients undergoing ophthalmic surgery. Anaesthesia 1996, 51, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Wang, H. Effect of propofol and sevoflurane on tumor killing activity of peripheral blood natural killer cells in patients with gastric cancers. J. Int. Med. Res. 2020, 48, 300060520904861. [Google Scholar] [CrossRef]
- Bai, Z.M.; Li, X.F.; Yang, Y.; Yang, Y.F.; Lv, D.R.; Tang, L.L. Propofol inhibited gastric cancer proliferation via the hsa-miR-328-3p/STAT3 pathway. Clin. Transl. Oncol. 2021, 23, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Tabnak, P.; Masrouri, S.; Geraylow, K.R.; Zarei, M.; Esmailpoor, Z.H. Targeting miRNAs with anesthetics in cancer: Current understanding and future perspectives. Biomed. Pharmacother. 2021, 144, 112309. [Google Scholar] [CrossRef]
- Kang, F.C.; Wang, S.C.; So, E.C.; Chang, M.M.; Wong, K.L.; Cheng, K.S.; Chen, Y.C.; Huang, B.M. Propofol may increase caspase and MAPK pathways, and suppress the Akt pathway to induce apoptosis in MA-10 mouse Leydig tumor cells. Oncol. Rep. 2019, 41, 3565–3574. [Google Scholar] [CrossRef]
- Yang, K.S.; Che, P.C.; Hsieh, M.J.; Lee, I.N.; Wu, Y.P.; Chen, M.S.; Chen, J.C. Propofol induces apoptosis and ameliorates 5-fluorouracil resistance in OSCC cells, by reducing the expression and secretion of amphiregulin. Mol. Med. Rep. 2022, 25, 36. [Google Scholar] [CrossRef]
- Lim, J.; Oh, C.; Yoon, T.; Lee, J.Y.; Lee, S.; Hoo, Y.; Yang, J.; Kim, S. The effect of propofol and sevoflurane on cancer cell, natural killer cell, and cytotoxic T lymphocyte function in patients undergoing breast cancer surgery: An in vitro analysis. BMC Cancer 2018, 15, 159. [Google Scholar] [CrossRef]
- van Breemen, M.S.; Rijsman, R.M.; Taphoorn, M.J.; Walchenbach, R.; Zwinkels, H.; Vecht, C.J. Efficacy of anti-epileptic drugs in patients with gliomas and seizures. J. Neurol. 2009, 256, 1519–1526. [Google Scholar] [CrossRef]
- Löscher, W. Effects of the antiepileptic drug valproate on metabolism and function of inhibitory and excitatory amino acid in the brain. Neurochem. Res. 1993, 18, 485–502. [Google Scholar] [CrossRef]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Sami, S.; Hoti, N.; Xu, H.M.; Shen, Z.; Huang, X. Valproic acid inhibits the growth of cervical cancer both in vitro and in vivo. J. Biochem. 2008, 144, 357–362. [Google Scholar] [CrossRef]
- Xia, Q.; Sung, J.; Chowdhury, W.; Chen, C.L.; Höti, N.; Shabbeer, S.; Carducci, M.; Rodriguez, R. Chronic administration of valproic acid inhibits prostate cancer cell growth in vitro and in vivo. Cancer Res. 2006, 66, 7237–7244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidana, A.; Wang, M.; Shabbeer, S.; Chowdhury, W.H.; Netto, G.; Lupold, S.E.; Carducci, M.; Rodriguez, R. Mechanism of growth inhibition of prostate cancer xenografts by valproic acid. J. Biomed. Biotechnol. 2012, 2012, 180363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Piao, J.; Li, N.; Yang, Y.; Kim, K.Y.; Lin, Z. Valproic acid targets HDAC1/2 and HDAC1/PTEN/Akt signalling to inhibit cell proliferation via the induction of autophagy in gastric cancer. FEBS J. 2020, 287, 2118–2133. [Google Scholar] [CrossRef]
- Gan, C.P.; Hamid, S.; Hor, S.Y.; Zain, R.B.; Ismail, S.M.; Wan Mustafa, W.M.; Teo, S.H.; Saunders, N.; Cheong, S.C. Valproic acid: Growth inhibition of head and neck cancer by induction of terminal differentiation and senescence. Head Neck 2012, 34, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [Green Version]
- Heers, H.; Stanislaw, J.; Harrelson, J.; Lee, M.W. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur. J. Pharmacol. 2018, 835, 61–74. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Liang, Q.; Sun, G. Valproic acid reverses sorafenib resistance through inhibiting activated Notch/Akt signaling pathway in hepatocellular carcinoma. Fundam. Clin. Pharmacol. 2021, 35, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Summers, N.; Vanderpuye-Orgle, J.; Reinhart, M.; Gallagher, M.; Sartor, O. Efficacy and safety of post-docetaxel therapies in metastatic castration-resistant prostate cancer: A systematic review of the literature. Curr. Med. Res. Opin. 2017, 33, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Tsaur, I.; Chun, F.K.; Bereiter-Hahn, J.; Blaheta, R.A. Influence of HDAC inhibitor valproic acid on the growth and proliferation of temsirolmus-resistant prostate cancer cells in vitro. Cancers 2019, 11, 566. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Guan, W. Valproic acid: A promising therapeutic agent in glioma treatment. Front. Oncol. 2021, 11, 687362. [Google Scholar] [CrossRef]
- Lipska, K.; Gumieniczek, A.; Filip, A.A. Anticonvulsant valproic acid and other short chain fatty acids as novel anticancer therapeutics: Possibilities and challenges. Acta Pharm. 2020, 70, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinari, N.; De Tursi, M.; Grassadonia, A.; Zilli, M.; Stuppia, L.; Iacobelli, S.; Natoli, C. An epigenetic approach to pancreatic cancer treatment: The prospective role of histone deacetylase inhibitors. Curr. Cancer Drug Targets 2012, 12, 439–452. [Google Scholar] [CrossRef]
- Hornig, E.; Heppt, M.V.; Graf, S.A.; Ruzicka, T.; Berking, C. Inhibition of histone deacetylases in melanoma-a perspective from bench to bedside. Exp. Dermatol. 2016, 25, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of valproic acid in combination with S-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014, 34, 5187–5192. [Google Scholar]
- Zhang, S.; Tang, Z.; Qing, B.; Tang, R.; Duan, Q.; Ding, S.; Deng, D. Valproic acid promotes the epithelial-to-mesenchymal transition of breast cancer cells through stabilization of Snail and transcriptional upregulation of Zeb1. Eur. J. Pharmacol. 2019, 865, 172745. [Google Scholar] [CrossRef]
- Chu, B.F.; Karpenko, M.J.; Liu, Z.; Aimiuwu, J.; Villalona-Calero, M.A.; Chan, K.K.; Grever, M.R.; Otterson, G.A. Phase I study of 5-aza-2′-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2013, 71, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Li, M.; Meng, F.; Yu, Z.; Chen, Y.; Cui, G. Combination of SB431542, CHIR99021 and PD0325901 has a synergic effect on abrogating valproic acid-induced epithelial-mesenchymal transition and stemness in HeLa, 5637 and SCC-15 cells. Oncol. Rep. 2019, 41, 3545–3554. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.N.; Pavone, K.J.; Naranjo, M. Multimodal general anesthesia: Theory and practice. Anesth. Analg. 2018, 127, 1246–1258. [Google Scholar] [CrossRef]
- Biki, B.; Mascha, E.; Moriarty, D.C.; Fitzpatrick, J.M.; Sessler, D.I.; Buggy, D.J. Anesthetic technique for radical prostatectomy surgery affects cancer recurrence: A retrospective analysis. Anesthesiology 2008, 109, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Christopherson, R.; James, K.E.; Tableman, M.; Marshall, P.; Johnson, F.E. Long-term survival after colon cancer surgery: A variation associated with choice of anesthesia. Anesth. Analg. 2008, 107, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; He, X.F.; Xu, Q.R.; Xu, Y.J.; Shen, J. Sevoflurane downregulates insulin-like growth factor-1 to inhibit cell proliferation, invasion and trigger apoptosis in glioma through the PI3K/AKT signaling pathway. Anticancer Drugs 2019, 30, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zheng, Y.; Rong, W. Sevoflurane induces apoptosis and inhibits the growth and motility of colon cancer in vitro and in vivo via inactivating Ras/Raf/MEK/ERK signaling. Life Sci. 2019, 239, 116916. [Google Scholar] [CrossRef]
- Fan, L.; Wu, Y.; Wang, J.; He, J.; Han, X. Sevoflurane inhibits the migration and invasion of colorectal cells through regulating ERK/MMP-9 pathway by up-regulating miR-203. Eur. J. Pharmacol. 2019, 850, 43–52. [Google Scholar] [CrossRef]
- Liu, J.; Yang, L.; Guo, X.; Jin, G.; Wang, Q.; Lv, D.; Liu, J.; Chen, Q.; Song, Q.; Li, B. Sevoflurane suppresses proliferation by upregulating microRNA-203 in breast cancer cells. Mol. Med. Rep. 2018, 18, 455–460. [Google Scholar] [CrossRef]
- Liang, H.; Yang, C.X.; Zhang, B.; Wang, H.B.; Liu, H.Z.; Lai, X.H.; Liao, M.J.; Zhang, T. Sevoflurane suppresses hypoxia-induced growth and metastasis of lung cancer cells via inhibiting hypoxia-inducible factor-1α. J. Anesth. 2015, 29, 821–830. [Google Scholar] [CrossRef]
- Hu, J.; Hu, J.; Jiao, H.; Li, Q. Anesthetic effects of isoflurane and the molecular mechanism underlying isoflurane-inhibited aggressiveness of hepatic carcinoma. Mol. Med. Rep. 2018, 18, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Stollings, L.M.; Jia, L.J.; Tang, P.; Dou, H.; Lu, B.; Xu, Y. Immune modulation by volatile anesthetics. Anesthesiology 2016, 125, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchi, C.F.; Mele, A.; Baliva, G.; Sciò, M.; Fucci, M.; Pasquini, P.; Corona, R. Prognostic value of anesthesia type for patients treated for cutaneous melanoma. Dermatol. Surg. 1995, 21, 786–788. [Google Scholar] [CrossRef] [PubMed]
- Cata, J.P.; Hagan, K.B.; Bhavsar, S.D.; Arunkumar, R.; Grasu, R.; Dang, A.; Carlson, R.; Arnold, B.; Potylchansky, Y.; Lipski, I.; et al. The use of isoflurane and desflurane as inhalational agents for glioblastoma surgery. A survival analysis. J. Clin. Neurosci. 2017, 35, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.Y.; Zhang, S.J.; Liu, L.; Chen, Q.S.; Yu, L.N.; Zhang, F.J.; Yan, M. Sevoflurane promotes the expansion of glioma stem cells through activation of hypoxia-inducible factors in vitro. Br. J. Anaesth. 2015, 114, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.C.; Shan, W.R.; Zhou, D.; Zeng, X.Q.; Zuo, K.; Pan, D.F.; Zeng, W.A.; Zuo, Z.Y. Sevoflurane promotes migration, invasion, and colony-forming ability of human glioblastoma cells possibly via increasing the expression of cell surface protein 44. Acta Pharmacol. Sin. 2019, 40, 1424–1435. [Google Scholar] [CrossRef]
- Lu, N.; Piao, M.H.; Feng, C.S.; Yuan, Y. Isoflurane promotes epithelial-to-mesenchymal transition and metastasis of bladder cancer cells through HIF-1α-β-catenin/Notch1 pathways. Life Sci. 2020, 258, 118154. [Google Scholar] [CrossRef]
- Zhang, W.; Xue, F.; Xie, S.; Chen, C.; Li, J.; Zhu, X. Isoflurane promotes proliferation of squamous cervical cancer cells through mTOR-histone deacetylase 6 pathway. Mol. Cell. Biochem. 2021, 476, 45–55. [Google Scholar] [CrossRef]
- Tazawa, K.; Koutogiannaki, S.; Chamberlain, M.; Yuki, K. The effect of different anesthetics on tumor cytotoxicity by natural killer cells. Toxicol. Lett. 2017, 266, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Buckley, A.; McQuaid, S.; Johnson, P.; Buggy, D.J. Effect of anaesthetic technique on the natural killer cell anti-tumour activity of serum from women undergoing breast cancer surgery: A pilot study. Br. J. Anaesth. 2014, 113 (Suppl. 1), i56–i62. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Status | Study Title | Condition | Interventions | Locations |
|---|---|---|---|---|
| Recruiting | The effect of ketamine on immune function and prognosis in patients undergoing colorectal cancer resection | Colorectal cancer | Drug: saline Drug: ketamine | Department of Anesthesiology and Pain Medicine, Yonsei University College of Medicine, Seoul, Republic of Korea, 03772 |
| Status | Study Title | Condition | Interventions | Locations |
|---|---|---|---|---|
| Recruiting | The effects of propofol-based intravenous vs. sevoflurane inhalation anaesthesia on inflammation and circulating tumor cells in paediatric tumor surgery—a pilot study | Solid tumor Carcinoma Malignancy Cancer | Drug: propofol Drug: sevoflurane | Hong Kong Children’s Hospital, Hong Kong, China. |
| Recruiting | The influence of anesthesia on postoperative outcome and complications in colorectal cancer patients | Colorectal cancer | Drug: TIVA + lidocaine Drug: sevoflurane + lidocaine Drug: TIVA + Placebo Drug: Sevoflarane + placebo | Clinical ATI, Str Crotoiilor nr 19-21, Clusnapoca, Cluj, Romania, 400162 |
| Recruiting | Contribution to the elucidation of the mechanisms and effects by which certain perianesthetic interventions modify long-term evolution of patients with digestive cancers subjected to surgery | Colorectal cancer | Drug: Lidocaine 1% injectable solution Biological: blood extraction Drug: sevoflurane Drug: propofol | Institutu Regional de Gastroenterologie si Hepatologi Cluj-Napoca, Cluj, Romania, 400469 |
| Year | Study Design | Cell Lines/Animal | Anesthetics | Pathways | Major Findings | Ref. |
|---|---|---|---|---|---|---|
| 2014 | In vitro | MA-10 mouse Leydig tumor cell line | Midazolam | Caspase cascade, p-Akt pathway, P-38, and JNK pathways | Midazolam induces MA-10 cell apoptosis via activation of the caspase cascade, the inhibition of p-Akt, P-38, and JNK pathways. | [31] |
| 2016 | In vitro | MA-10 mouse Leydig tumor cell line | Midazolam | Apoptosis pathway, autophagy, and ER stress | Midazolam induces apoptosis of MA-10 cells through the induction of ER stress, regulation of cell cycle, and autophagy. | [32] |
| 2018 | In vitro | Human A549 non-small cell lung carcinoma | Midazolam | Mitochondria intrinsic apoptosis pathway, miR-520d-5p, STAT3 pathway | Midazolam induces apoptosis of A549 cells via induction of miR-520d-5p-inhibited STAT3 signaling. | [33] |
| 2013 | In vitro and xenograft model | Human K562 leukemia cells and HT29 colon cancer cells | Midazolam | Cell cycle, Intrinsic apoptosis pathway, ERK pathway, reactive oxygen species | Midazolam inhibits proliferation and induces mitochondria intrinsic apoptotic signaling. Midazolam also inhibits HT29 tumor growth in a mouse xenograft model. The mechanisms involve the inhibition of ROS, induction of apoptosis, and inhibition of growth-related proteins. | [34] |
| 2020 | In vitro | Human hepatocellular carcinoma cell, HepG2 | Midazolam | Cell migration, invasion, proliferation, apoptosis | Midazolam inhibits cell proliferation and promotes apoptosis by inducing microRNA miR-124-3p | [35] |
| 2010 | In vitro | Human Jurkat T lymphoma cell, SHEP neuroblastoma cell, and primary rat cortex neurons | Midazolam | The mitochondrial intrinsic apoptotic pathway | Midazolam induces apoptosis of all cell types through the induction of caspase 9 and suppression of BCL-2, whereas deficiency of FADD and caspase 8 has no effect. | [36] |
| 2021 | In vitro | Human glioblastoma cell lines U118-MG andU87MG | Remimazolan | Apoptotic pathway, NF-κB pathway | Remimazolan induces apoptosis of glioblastoma cells through the activation of caspase cascade, inhibition of NF-κB, and downstream anti-apoptotic protein, such as XIAP and survivin. | [38] |
| 2018 | In vitro and xenograft model | Human A549 non-small cell lung carcinoma, H4 neuroglioma cell line | Midazolam, Dexmedetomidine | The mitochondrial intrinsic apoptotic pathway, PBR pathway, cell migration | Dexmedetomidine promotes cancer progression in both cell lines, whereas midazolam inhibits cancer progression by induction of mitochondrial intrinsic apoptotic pathway. The inhibitory effect of midazolam is partly mediated by PBR. | [39] |
| 2021 | In vitro | Human A549 non-small cell lung carcinoma, breast cancer cell line MCF-7, MDA-MB-231 human breast cancer cell line | Midazolam | Cell migration, invasion, epithelial-mesenchymal transition (EMT), PBR | At low dosage, midazolam treatment inhibits TGF-β-induced proliferation, migration, invasion, and EMT in A549 and MCF-7 through PBR. Midazolam inhibits proliferation, migration, invasion, and mesenchymal marker of MDA-MB-231 cells. | [40] |
| 2020 | In vivo | Hyperglycemia-induced pulmonary vascular leakage and cancer metastasis in diabetic mice | Midazolam | Lung metastasis, reactive oxygen species, endothelium leakage | Subcutaneous injection of midazolam inhibited hyperglycemia-induced cancer metastasis in the lungs of diabetic mice by preventing the generation of ROS, activation of transglutaminase, and subsequent vascular leakage. | [41] |
| 2021 | In vitro and xenograft model | Cisplatin-resistant non-small cell lung cancer cell line | Midazolam | Proliferation, apoptosis, microRNA | Midazolam suppresses cell proliferation and viability and promotes cell apoptosis in cisplatin-treated CR-NSCLC cells. Midazolam enhances cisplatin sensitivity in CR-NSCLC cells via modulating the miR-194-5p/hook microtubule-tethering protein 3 (HOOK3) axis. | [42] |
| 2022 | In vitro and transgenic mouse model | Murine pancreatic ductal adenocarcinoma cell lie, transgenic PDAC mouse model (LSLkrasG12D/+; Trp53flox/flox;Pdx-1cre/+ [KPPC]) | Midazolam | Anti-tumor, inflammatory response | Midazolam significantly inhibited tumor size and proliferative index of Ki-67 and cyclins in PDAC through PBR. Midazolam inhibits cancer-associated neutrophils, macrophages, PMN, and pro-inflammatory cytokines through PBR. | [45] |
| 2012 | In vitro | human neuroblastoma cell line SN-K-SH | Haloperidol | Apoptotic pathway | Haloperidol, but not risperidone and paliperidone, induces neuroblastoma cell death via induction of apoptosis | [48] |
| 2020 | In vitro | Human glioblastoma (GBM) U87, U251, and T98 cell lines | Haloperidol | Cell cycle, cell migration, apoptotic pathway | Treatment with haloperidol reduces the viability of these GBM cell lines by induction of apoptosis. Haloperidol inhibits cell migration and CD24/CD44 alteration expression. Haloperidol, combined with TMZ and radiation therapies, further increased tumor cell death. | [51] |
| 2009 | In vitro | PC12 rat pre-neuronal cell line | Haloperidol | Akt pathway, mitochondria intrinsic apoptotic pathway | Haloperidol inactivates Akt, which induces the dephosphorylation of serine in Bcl-XS and promotes its association with the mitochondrial voltage-dependent anion channel (VDAC), and with cytochrome c- and caspase-3-dependent events. | [52] |
| 2020 | In vitro | human ovarian cancer cell lines OVCAR-3, SKOV3, A2780, 3AO, COC1, OV-90 | Ketamine | Cell proliferation, apoptotic pathway, long noncoding RNA, histone acetylation, and methylation | Ketamine inhibits the proliferation and survival of six ovarian cancer cell lines by regulating P300-mediated H3K27 acetylation activation in the promoter of PVT1, which in turn binds EZH2 and promotes p57 expression. | [62] |
| 2021 | In vitro and xenograft model | Human breast cancer cell lines MCF-7 and T47D; Human liver cancer cell (HCC) lines HepG2 and Huh7 | Ketamine | Apoptosis, ferroptosis, long noncoding RNA (lncRNA), reactive oxygen species | Ketamine suppresses the viability and proliferation of liver and breast cancer cells through the activation of ferroptosis. Ketamine-induced ferroptosis is mediated by the inhibition of lncPVT1 and glutathione peroxidase 4 (GP4) in HCC, whereas ketamine induces the levels of MDA, lipid ROS, and Fe2+ and attenuates the KAT5-mediated decrease of GP4 in breast cancer cells. | [64,65] |
| 2016 | In vitro | Human ovarian carcinoma (SKOV-3) and prostate carcinoma (PC-3) | Bupivacaine | glycogen synthase kinase-3β, apoptosis | Bupivacaine reduces cell viability and inhibits proliferation and migration but induces apoptosis of both cell lines. Bupivacaine increased the phosphorylation of GSK-3βTyr216 in SKOV-3, which promotes the sensitivity of ovarian cancer cells to bupivacaine-induced cytotoxicity. | [71] |
| 2021 | In vitro | Human ovarian cancer cell line SKOV3 and COC1 | Lidocaine | Proliferation, apoptosis, Wnt pathway | Lidocaine inhibits proliferation, migration, and invasion, and induces apoptosis in ovarian cancer cell lines. Overexpression of Wnt/β-catenin signaling overcomes lidocaine-inhibited cell migration and invasion. | [72] |
| 2017 | In vitro and xenograft model | Human liver cancer cell (HCC) lines HepG2 | Lidocaine | Cell cycle, mitochondria intrinsic apoptotic pathway, MAPK pathways | Lidocaine inhibits the growth but stimulates apoptosis of HepG2 through ERK/P38-mediated mitochondria intrinsic apoptotic pathway. | [73] |
| 2018 | In vitro | Human prostate cancer cell line DU-145 | Levobupivacaine | Glycolysis, oxidative phosphorylation, reactive oxygen species, cell cycle | Levobupivacaine induces inhibition of glycolysis and oxidative phosphorylation in cancer cells, which in turn decreases cellular ATP production and consecutive bioenergetic crisis, together with reactive oxygen species generation. Cancer cells are arrested in the S phase without triggering apoptosis. | [74] |
| 2021 | In vitro and xenograft model | Human non-small cell lung cancer cell lines, A549 and A427 | Levobupivacaine | Ferroptosis, proliferation, apoptosis, reactive oxygen species, P53 | Treatment of levobupivacaine increases the levels of ROS, iron, Fe2+, and ferroptosis but attenuates migration and invasion of NSCLS. The levobupivacaine-induced ferroptosis is mediated by the regulation of P53. | [75] |
| 2021 | In vitro and xenograft model | Human gastric cancer cell lines, HGC27 and SGC7901 | Levobupivacaine | Ferroptosis, proliferation, microRNA | Treatment of levobupivacaine increases the levels of Fe2+/iron and lipid ROS and ferroptosis in erastin and RSL3-stimulated gastric cancer cells. This levobupivacaine-induced ferroptosis is mediated by upregulation of miR-489-3p and then targeting SLC7A11. | [76] |
| 2003 | In vitro | Human breast cancer cell line MCF-7 | Procaine | DNA methylation, proliferation | Procaine can demethylate densely hypermethylated CpG islands, restoring gene expression of epigenetically silenced genes and inhibiting cancer proliferation. | [77] |
| 2021 | In vitro | Human gastric cancer cell line SGC-7901 | Propofol | Proliferation, microRNA | Propofol inhibits the proliferation of gastric cancer cells by upregulating the has-miR-328-3p, which then downregulates the downstream genes, such as STAT3, MMP2, CCND1, and COX2. | [86] |
| 2019 | In vitro | MA-10 mouse Leydig tumor cell line | Propofol | Proliferation, mitochondria intrinsic apoptosis pathway, MAPK pathways, Akt pathways | Propofol decreases cell viability and increases mitochondria intrinsic apoptosis pathway. This apoptotic induction may be regulated by the MAPK activation and inhibition of Akt phosphorylation. | [88] |
| 2021 | In vitro | Human oral squamous cell carcinoma cell lines, SAS, SCC9 | Propofol | Apoptosis, drug- resistance, growth factors | Propofol decreases cell viability and promotes cell apoptosis. The expression and activation of amphiregulin is related to 5-FU resistance, where propofol ameliorates 5-FU drug resistance by downregulation of amphiregulin. | [89] |
| 2008 | In vitro and xenograft model | Human cervical cancer cell lines HeLa, Ca Ski and SiHa | Valproic acid (VPA) | Proliferation, histone acetylation, angiogenesis | VPA induces histone H3 acetylation and upregulates p21, cytostatic effects both in vitro and in vivo. VPA can also inhibit angiogenesis in vivo. | [94] |
| 2006, 2012 | In vitro and xenograft model | Human prostate cancer cell lines LNCaP, PC3, and DU145 | Valproic acid (VPA) | Proliferation, histone acetylation, cell cycle, apoptosis | Chronic administration of VPA decreases prostate cancer cell net proliferation with increased caspase activation both in vitro and in vivo. | [95,96] |
| 2020 | In vitro, xenograft model, and human samples | Human gastric cancer cell line SGC-7901 | Valproic acid (VPA) | Autophagy, apoptosis, histone deacetylase (HDAC), Akt pathways | Treatment of VPA inhibits HDAC1/2 activity and induced autophagy and then apoptosis. HDAC1/PTEN/Akt pathway and the regulation of BCL-2 and beclin-1 are involved in the inhibitory effects of VPA. The expression of HDAC correlates with poor prognosis in human gastric patients. | [97] |
| 2015 | In vitro | Human breast cancer cell lines, MCF-7 and SKBR3, MDA-MB-231, and BT474 | Valproic acid (VPA) | Proliferation, histone acetylation, heat shock protein, protein acetylation | Treatment of VPA inhibits proliferation of four breast cancer cell lines and with better inhibition in HER2-overexpressing SKBR3. VPA can also upregulate expression of p21 WAF1, cleaved caspase-3, and acetylation of heat shock protein 70. | [99] |
| 2020 | In vitro | Human sorafenib-resistant hepatocellular carcinoma cell line, HepG2-SR | Valproic acid (VPA), sorafenib | Drug resistance, Notch pathway, Akt pathway | Notch1 and Akt are upregulated in sorafenib-resistant cells. The combination of VPA and sorafenib treatment enhances sensitivity of drug-resistant cells and reverse the increased levels of Notch1 and Akt in HepG2-SR. | [101] |
| 2019 | In vitro | human prostate tumor cell lines PC3, DU-145, and LNCaP | Valproic acid (VPA) | Drug resistance, Akt-mTOR pathway, cell cycle, histone acetylation | In Temsirolimus resistant cell lines, the elevation of cell proliferation and clonal growth is associated with cell cycling proteins cdk1 and cyclin B, along with increases in Akt-mTOR signaling but decreases in p19, p21, and p27. Treatment of VPA inhibits cell growth and upregulates the acetylated histones H3 and H4 together with the decrease of Cdk1 and cyclin B phosphorylation of mTOR and the mTOR sub-complex Raptor. | [104] |
| 2019 | In vitro | Human glioblastoma cell lines, U251 and U87 | Sevoflurane | Proliferation, invasion and migration, apoptosis, insulin-like growth factor (IGF-1) pathway, PI3K/Akt pathway. | Sevoflurane treatment inhibits proliferation, migration, and invasion but promotes apoptosis in glioblastoma cell lines. This inhibitory effect of sevoflurane is mediated by IGF-1/PI3K/Akt signaling. | [116] |
| 2019 | In vitro and xenograft model | Human colon cancer cell line SW480 and SW620 | Sevoflurane | Proliferation, cell cycle, apoptosis, autophagy, invasion, and epithelial-mesenchymal transition | Sevoflurane treatment inhibits proliferation, invasion, and cell cycle progression, and promotes apoptosis and autophagy through Raf/MEK/ERK pathways. | [117] |
| 2019 | In vitro, human samples | Human colorectal cancer (CRC) cell lines SW620 and HCT116 | Sevoflurane | Migration and invasion, ERK/MMP pathway, microRNA | Sevoflurane treatment inhibits migration and invasion but not the proliferation of CRC cell lines. Treatment of sevoflurane downregulates phosphorylation of ERK (p-ERK) but restores expression of miR-203. Inhibition of miR-203 attenuates the inhibitory effect of sevoflurane on cell migration, invasion, and p-ERK. | [118] |
| 2018 | In vitro | Human breast cancer cell lines, MCF-7 and MDA-MB-231 | Sevoflurane | Proliferation, cell cycle, microRNA | Treatment of sevoflurane inhibits the proliferation of breast cancer cell lines by activating miR-203. | [119] |
| 2015 | In vitro | Human non-small cell lung carcinoma cell line, A549 | Sevoflurane | Hypoxia, proliferation, metastasis, P38 MAPK | Sevoflurane treatment suppresses hypoxia-induced proliferation and metastasis of A549 cells by modulating HIF-1α and its downstream genes. In addition, the p38 MAPK pathway is involved in regulating HIF-1α by sevoflurane. | [120] |
| 2018 | In vitro, xenograft model, and human samples | Primary culture of human hepatocellular carcinoma | Isoflurane | Proliferation, apoptosis, migration and invasion, PI3K/Akt pathway, NF-κB pathway | Treatment of isoflurane inhibits growth and decreased viability of liver cancer cells in vitro and in vivo, and the apoptotic rate is increased in cells obtained from isoflurane-treated patients. Treatment of isoflurane inhibits PI3K/Akt to regulate cell survival, whereas isoflurane-attenuated NF-κB inhibits migration and invasion of cancer cells. | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.-C.; Liao, K.-S.; Yeh, L.-R.; Wang, Y.-K. Drug Repurposing: The Mechanisms and Signaling Pathways of Anti-Cancer Effects of Anesthetics. Biomedicines 2022, 10, 1589. https://doi.org/10.3390/biomedicines10071589
Wu K-C, Liao K-S, Yeh L-R, Wang Y-K. Drug Repurposing: The Mechanisms and Signaling Pathways of Anti-Cancer Effects of Anesthetics. Biomedicines. 2022; 10(7):1589. https://doi.org/10.3390/biomedicines10071589
Chicago/Turabian StyleWu, King-Chuen, Kai-Sheng Liao, Li-Ren Yeh, and Yang-Kao Wang. 2022. "Drug Repurposing: The Mechanisms and Signaling Pathways of Anti-Cancer Effects of Anesthetics" Biomedicines 10, no. 7: 1589. https://doi.org/10.3390/biomedicines10071589
APA StyleWu, K. -C., Liao, K. -S., Yeh, L. -R., & Wang, Y. -K. (2022). Drug Repurposing: The Mechanisms and Signaling Pathways of Anti-Cancer Effects of Anesthetics. Biomedicines, 10(7), 1589. https://doi.org/10.3390/biomedicines10071589