
Molecular Mechanisms of Autophagy in Cancer Development, Progression, and Therapy

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Biological Mechanisms of Autophagy
3. Transcriptional Regulation of Autophagy
4. Autophagy and Anoikis
5. Autophagy and Apoptosis
6. Autophagy and Ferroptosis
7. Autophagy and Its Dual Role in the Regulation of Cancer
7.1. Tumor Suppression
7.2. Tumor Promotion
8. Autophagy in Specific Tumor Types
8.1. Breast Cancer
8.2. Lung Cancer
8.3. Mesothelioma
8.4. Pancreatic Cancer
9. Targeting Autophagy as Cancer Treatment
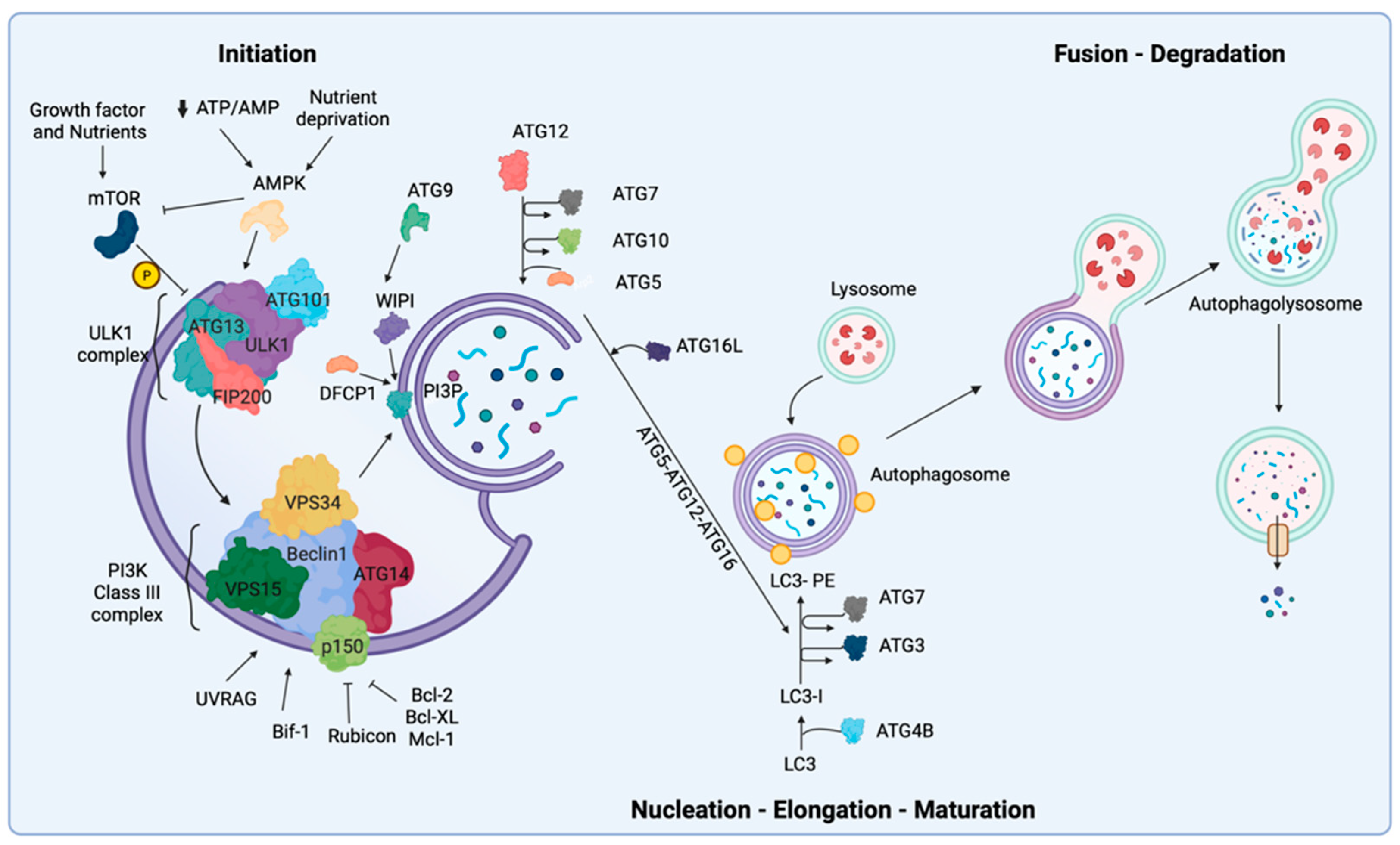
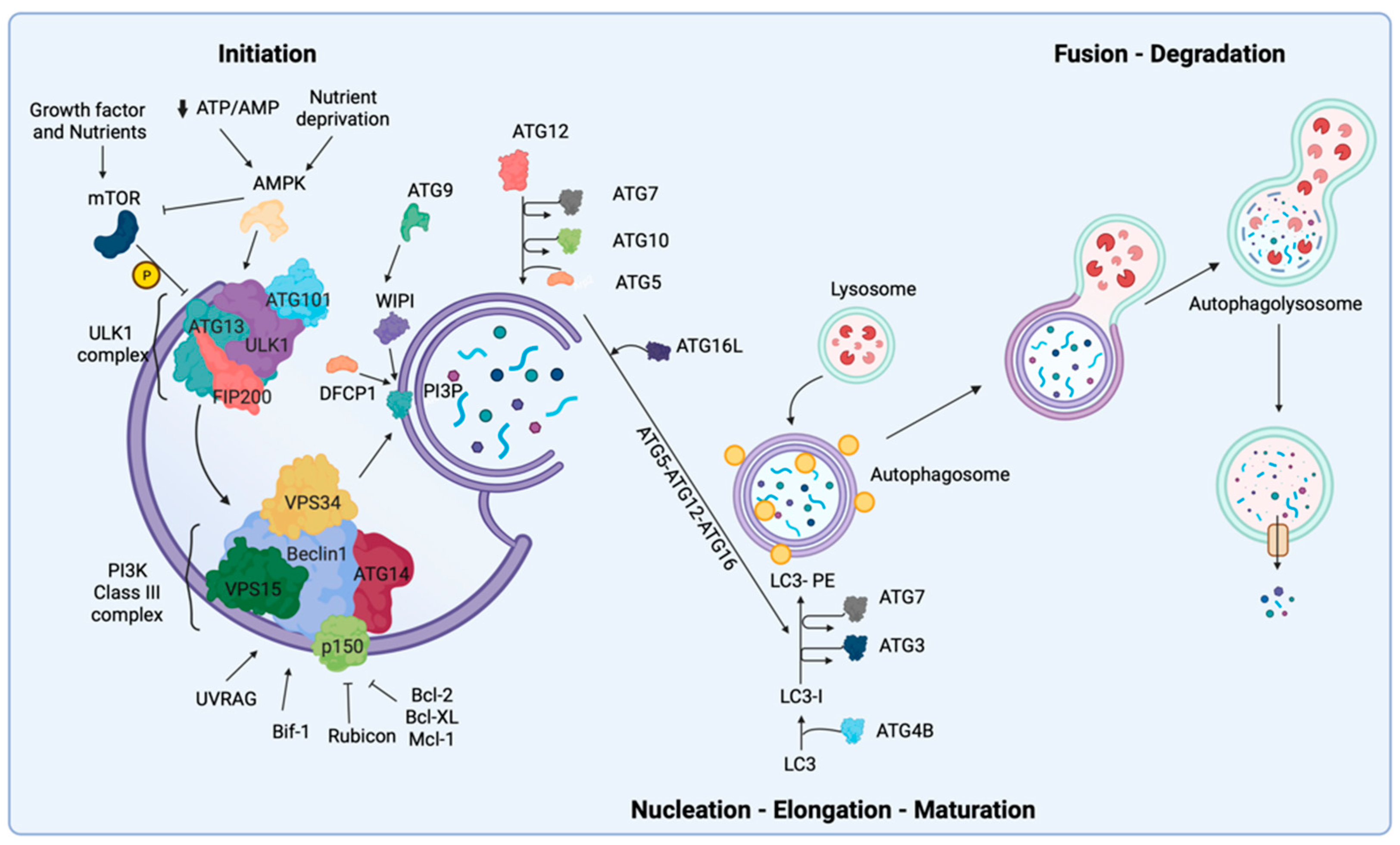
9.1. Initiation and Nucleation Phases
9.2. Elongation and Closure
9.3. Vesicle Degradation
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Kamada, Y.; Kabeya, Y.; Sekito, T.; Ohsumi, Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell 2008, 19, 2039–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Hong, Y. The fluorescence toolbox for visualizing autophagy. Chem. Soc. Rev. 2020, 49, 8354–8389. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafa, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Bonora, M.; Ingusci, S.; Previati, M.; Marchi, S.; Zucchini, S.; Perrone, M.; Wieckowski, M.R.; Castellazzi, M.; Pugliatti, M.; et al. Antipsychotic drugs counteract autophagy and mitophagy in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020078118. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Han, H.; Wu, L.; Pan, B.; Dong, B.; Yin, C.C.; Tian, Z.; Liu, X.; Yang, Y.; Zhang, H.; et al. iASPP facilitates tumor growth by promoting mTOR-dependent autophagy in human non-small-cell lung cancer. Cell Death Dis. 2017, 8, e3150. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Fairlie, W.D.; Tran, S.; Lee, E.F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 115–158. [Google Scholar] [CrossRef]
- Fulda, S. Targeting autophagy for the treatment of cancer. Biol. Chem. 2018, 399, 673–677. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Lee, M.S. Autophagy-a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Z.; Shu, S.; Cai, J.; Tang, C.; Dong, Z. AMPK/mTOR Signaling in Autophagy Regulation During Cisplatin-Induced Acute Kidney Injury. Front. Physiol. 2020, 11, 619730. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife 2016, 5, e11058. [Google Scholar] [CrossRef]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Jiang, S.; Li, T.; Yang, Z.; Yi, W.; Di, S.; Sun, Y.; Wang, D.; Yang, Y. AMPK orchestrates an elaborate cascade protecting tissue from fibrosis and aging. Ageing Res. Rev. 2017, 38, 18–27. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachari, M.; Longo, M.; Ganley, I.G. Aberrant autophagosome formation occurs upon small molecule inhibition of ULK1 kinase activity. Life Sci. Alliance 2020, 3, e202000815. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Fujioka, Y.; Suzuki, S.W.; Noshiro, D.; Suzuki, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ando, T.; Noda, N.N.; et al. The Intrinsically Disordered Protein Atg13 Mediates Supramolecular Assembly of Autophagy Initiation Complexes. Dev. Cell 2016, 38, 86–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotani, T.; Kirisako, H.; Koizumi, M.; Ohsumi, Y.; Nakatogawa, H. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc. Natl. Acad. Sci. USA 2018, 115, 10363–10368. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 Kinase Domain Dynamics Regulate the Autophagic PI 3-Kinase Complex. Mol. Cell 2017, 67, 528–534.e523. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papinski, D.; Schuschnig, M.; Reiter, W.; Wilhelm, L.; Barnes, C.A.; Maiolica, A.; Hansmann, I.; Pfaffenwimmer, T.; Kijanska, M.; Stoffel, I.; et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell 2014, 53, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Noda, N.N.; Inagaki, F. Mechanisms of Autophagy. Annu. Rev. Biophys. 2015, 44, 101–122. [Google Scholar] [CrossRef]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Shen, H.M.; Mizushima, N. At the end of the autophagic road: An emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 2014, 39, 61–71. [Google Scholar] [CrossRef]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemesath, T.J.; Steingrimsson, E.; McGill, G.; Hansen, M.J.; Vaught, J.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A.; Fisher, D.E. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994, 8, 2770–2780. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong, A.L.; Patange, S.; Raben, N.; Puertollano, R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014, 7, ra9. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013, 200, 475–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiacchiera, F.; Matrone, A.; Ferrari, E.; Ingravallo, G.; Lo Sasso, G.; Murzilli, S.; Petruzzelli, M.; Salvatore, L.; Moschetta, A.; Simone, C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ. 2009, 16, 1203–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.; Tao, R.; DePinho, R.A.; Dong, X.C. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem. 2012, 287, 39107–39114. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Seok, S.; Fu, T.; Choi, S.E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gang, H.; Dhingra, R.; Wang, Y.; Mughal, W.; Gordon, J.W.; Kirshenbaum, L.A. Epigenetic regulation of E2F-1-dependent Bnip3 transcription and cell death by nuclear factor-kappaB and histone deacetylase-1. Pediatr. Cardiol. 2011, 32, 263–266. [Google Scholar] [CrossRef]
- Boyd, J.M.; Malstrom, S.; Subramanian, T.; Venkatesh, L.K.; Schaeper, U.; Elangovan, B.; D’Sa-Eipper, C.; Chinnadurai, G. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994, 79, 341–351. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Polager, S.; Ofir, M.; Ginsberg, D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008, 27, 4860–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.; Yurkova, N.; Zhang, T.; Gang, H.; Aguilar, F.; Weidman, D.; Scramstad, C.; Weisman, H.; Kirshenbaum, L.A. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc. Natl. Acad. Sci. USA 2008, 105, 20734–20739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Jang, Y.; You-Ten, A.I.; Okada, H.; Liepa, J.; Wakeham, A.; Zaugg, K.; Mak, T.W. p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc. Natl. Acad. Sci. USA 2004, 101, 14057–14062. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Ma, Q.; Chen, L.; Li, P.; Zhang, M.; Ramamoorthy, S.; Nawaz, Z.; Shimojima, T.; Wang, H.; Yang, Y.; et al. MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J. Biol. Chem. 2009, 284, 13987–14000. [Google Scholar] [CrossRef] [Green Version]
- Renault, V.M.; Thekkat, P.U.; Hoang, K.L.; White, J.L.; Brady, C.A.; Kenzelmann Broz, D.; Venturelli, O.S.; Johnson, T.M.; Oskoui, P.R.; Xuan, Z.; et al. The pro-longevity gene FoxO3 is a direct target of the p53 tumor suppressor. Oncogene 2011, 30, 3207–3221. [Google Scholar] [CrossRef] [Green Version]
- Brady, O.A.; Jeong, E.; Martina, J.A.; Pirooznia, M.; Tunc, I.; Puertollano, R. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. eLife 2018, 7, e40856. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenific, C.M.; Wittmann, T.; Debnath, J. Autophagy in adhesion and migration. J. Cell Sci. 2016, 129, 3685–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Debnath, J.; Mills, K.R.; Collins, N.L.; Reginato, M.J.; Muthuswamy, S.K.; Brugge, J.S. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002, 111, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, T.; Wei, Q.; Zhang, Y.; Jia, X.; Wan, Z.; Han, L. Upregulation of BNIP3 mediated by ERK/HIF-1alpha pathway induces autophagy and contributes to anoikis resistance of hepatocellular carcinoma cells. Future Oncol. 2014, 10, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Sossey-Alaoui, K.; Thompson, C.L.; Danielpour, D.; Schiemann, W.P. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 150–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; Fan, J.; et al. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 2018, 412, 108–117. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.; Liu, H.; Jiang, S.; Wang, G.; Sun, L.; Li, J.; Wang, X.; Yu, S.; Huang, J.; et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis. 2020, 11, 198. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Eritja, N.; Lock, R.; Debnath, J. Autophagy restricts proliferation driven by oncogenic phosphatidylinositol 3-kinase in three-dimensional culture. Oncogene 2013, 32, 2543–2554. [Google Scholar] [CrossRef] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Nagasaka, A.; Kawane, K.; Yoshida, H.; Nagata, S. Apaf-1-independent programmed cell death in mouse development. Cell Death Differ. 2010, 17, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Burgess, D.J. Apoptosis: Refined and lethal. Nat. Rev. Cancer 2013, 13, 79. [Google Scholar] [CrossRef]
- Chen, C.W.; Wu, M.S.; Huang, Y.J.; Lin, P.W.; Shih, C.J.; Lin, F.P.; Chang, C.Y. Iridovirus CARD Protein Inhibits Apoptosis through Intrinsic and Extrinsic Pathways. PLoS ONE 2015, 10, e0129071. [Google Scholar] [CrossRef]
- Kim, M.J.; Kwon, S.B.; Ham, S.H.; Jeong, E.S.; Choi, Y.K.; Choi, K.D.; Hong, J.T.; Jung, S.H.; Yoon, D.Y. H9 Inhibits Tumor Growth and Induces Apoptosis via Intrinsic and Extrinsic Signaling Pathway in Human Non-Small Cell Lung Cancer Xenografts. J. Microbiol. Biotechnol. 2015, 25, 648–657. [Google Scholar] [CrossRef]
- Verbrugge, I.; Johnstone, R.W.; Smyth, M.J. SnapShot: Extrinsic apoptosis pathways. Cell 2010, 143, 1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life 2022, 12, 256. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Selimovic, D.; Porzig, B.B.; El-Khattouti, A.; Badura, H.E.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef]
- Beaudouin, J.; Liesche, C.; Aschenbrenner, S.; Horner, M.; Eils, R. Caspase-8 cleaves its substrates from the plasma membrane upon CD95-induced apoptosis. Cell Death Differ. 2013, 20, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, J.M.; Dowdle, W.E.; DeJesus, R.; Wang, Z.; Bergman, P.; Kobylarz, M.; Lindeman, A.; Xavier, R.J.; McAllister, G.; Nyfeler, B.; et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017, 20, 2341–2356. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Gableske, S.; Quiles del Rey, M.; Malachowska, B.; Jedrychowski, M.P.; Biancur, D.E.; Schmidt, P.J.; Fleming, M.D.; Fendler, W.; Harper, J.W.; et al. NCOA4 maintains murine erythropoiesis via cell autonomous and non-autonomous mechanisms. Haematologica 2019, 104, 1342–1354. [Google Scholar] [CrossRef] [Green Version]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; Lenoel, I.; Stys, P.K.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 9327–9341. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Ryan, T.M.; Bush, A.I.; Masters, C.L.; Duce, J.A. The role of metallobiology and amyloid-beta peptides in Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. 1), 149–166. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.L.; Buchanan, D.D.; Ahmed, I.; Taylor, K.D.; Loriot, M.A.; Sinsheimer, J.S.; Bronstein, J.M.; Elbaz, A.; Mellick, G.D.; Rotter, J.I.; et al. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol. Dis. 2014, 62, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, Y.; Wu, J.; Zhang, Y.; Abdihamid, O.; Duan, C.; Li, B. Low expression of ferritinophagy-related NCOA4 gene in relation to unfavorable outcome and defective immune cells infiltration in clear cell renal carcinoma. BMC Cancer 2021, 21, 18. [Google Scholar] [CrossRef]
- Sun, Y.; Li, C.; Feng, J.; Li, Y.; Zhai, X.; Zhang, L.; Li, C. Ferritinophagic Flux Activation in CT26 Cells Contributed to EMT Inhibition Induced by a Novel Iron Chelator, DpdtpA. Oxidative Med. Cell. Longev. 2019, 2019, 8753413. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Di, J.; Cao, H.; Bai, J.; Zheng, J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015, 363, 101–107. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Wijshake, T.; Zou, Z.; Chen, B.; Zhong, L.; Xiao, G.; Xie, Y.; Doench, J.G.; Bennett, L.; Levine, B. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. USA 2021, 118, e2020478118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Cai, Y.; Xu, K.; Ren, X.; Sun, J.; Lu, S.; Chen, J.; Xu, P. Beclin1 overexpression suppresses tumor cell proliferation and survival via an autophagydependent pathway in human synovial sarcoma cells. Oncol. Rep. 2018, 40, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, S.; Gu, T.; Song, F.; Xue, Y.; Sun, D. MiR-140-3p Impedes Gastric Cancer Progression and Metastasis by Regulating BCL2/BECN1-Mediated Autophagy. OncoTargets Ther. 2021, 14, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.Y.; Byun, S.; Bae, S.H.; Min, D.S.; Woo, H.A.; Lee, K. TPT1 (tumor protein, translationally-controlled 1) negatively regulates autophagy through the BECN1 interactome and an MTORC1-mediated pathway. Autophagy 2017, 13, 820–833. [Google Scholar] [CrossRef] [Green Version]
- Komata, T.; Kanzawa, T.; Nashimoto, T.; Aoki, H.; Endo, S.; Nameta, M.; Takahashi, H.; Yamamoto, T.; Kondo, S.; Tanaka, R. Mild heat shock induces autophagic growth arrest, but not apoptosis in U251-MG and U87-MG human malignant glioma cells. J. Neuro-Oncol. 2004, 68, 101–111. [Google Scholar] [CrossRef]
- Marsh, T.; Kenific, C.M.; Suresh, D.; Gonzalez, H.; Shamir, E.R.; Mei, W.; Tankka, A.; Leidal, A.M.; Kalavacherla, S.; Woo, K.; et al. Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Dev. Cell 2020, 52, 591–604.e596. [Google Scholar] [CrossRef]
- Ambrosio, S.; Majello, B. Autophagy Roles in Genome Maintenance. Cancers 2020, 12, 1793. [Google Scholar] [CrossRef]
- Qiang, L.; Zhao, B.; Shah, P.; Sample, A.; Yang, S.; He, Y.Y. Autophagy positively regulates DNA damage recognition by nucleotide excision repair. Autophagy 2016, 12, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Qiang, L.; Wu, C.; Ming, M.; Viollet, B.; He, Y.Y. Autophagy controls p38 activation to promote cell survival under genotoxic stress. J. Biol. Chem. 2013, 288, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Umar, S.A.; Tanveer, M.A.; Nazir, L.A.; Divya, G.; Vishwakarma, R.A.; Tasduq, S.A. Glycyrrhizic Acid Prevents Oxidative Stress Mediated DNA Damage Response through Modulation of Autophagy in Ultraviolet-B-Irradiated Human Primary Dermal Fibroblasts. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 53, 242–257. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Missiroli, S.; Morciano, G.; Perrone, M.; Mantovani, C.M.; Anania, G.; Fiorica, F.; Pinton, P.; Giorgi, C. Understanding the Role of Autophagy in Cancer Formation and Progression Is a Real Opportunity to Treat and Cure Human Cancers. Cancers 2021, 13, 5622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, C.; Yang, P.; Li, C.; Li, M. Eldecalcitol induces apoptosis and autophagy in human osteosarcoma MG-63 cells by accumulating ROS to suppress the PI3K/Akt/mTOR signaling pathway. Cell. Signal. 2021, 78, 109841. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Xie, M.; Zhao, F.; Li, J.; Fan, C.; Zheng, H.; Wei, Z.; Ci, X.; Zhang, S. Daphnetin triggers ROS-induced cell death and induces cytoprotective autophagy by modulating the AMPK/Akt/mTOR pathway in ovarian cancer. Phytomedicine 2021, 82, 153465. [Google Scholar] [CrossRef]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Guzzo, S.; Mangolini, A.; dell’Atti, L.; Pinton, P.; Aguiari, G. The induction of AMPK-dependent autophagy leads to P53 degradation and affects cell growth and migration in kidney cancer cells. Exp. Cell Res. 2020, 395, 112190. [Google Scholar] [CrossRef]
- Zheng, H.C.; Zhao, S.; Xue, H.; Zhao, E.H.; Jiang, H.M.; Hao, C.L. The Roles of Beclin 1 Expression in Gastric Cancer: A Marker for Carcinogenesis, Aggressive Behaviors and Favorable Prognosis, and a Target of Gene Therapy. Front. Oncol. 2020, 10, 613679. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Li, G.; Huang, C.; Hou, Z.; Yang, X.; Luo, X.; Feng, Y.; Wang, G.; Hu, J.; Cao, Z. The autophagy-independent role of BECN1 in colorectal cancer metastasis through regulating STAT3 signaling pathway activation. Cell Death Dis. 2020, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Holah, N.S.; El-Dien, M.M.S.; Mahmoud, S.F. Expression of Autophagy Markers Beclin1 and LC3B in Prostatic Carcinoma: An Immunohistochemical Case-Control Study. Iran. J. Pathol. 2022, 17, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ling, L.; Zheng, Z.; Zhang, Y.; Wang, R.; Wu, M.; Zhang, N.; Hu, M.; Yang, X. ATG7-dependent and independent autophagy determine the type of treatment in lung cancer. Pharmacol. Res. 2021, 163, 105324. [Google Scholar] [CrossRef]
- Gorgulu, K.; Diakopoulos, K.N.; Ai, J.; Schoeps, B.; Kabacaoglu, D.; Karpathaki, A.F.; Ciecielski, K.J.; Kaya-Aksoy, E.; Ruess, D.A.; Berninger, A.; et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 2019, 156, 203–217.e220. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Wang, W.; Crespo, J.; Kryczek, I.; Li, W.; Wei, S.; Bian, Z.; Maj, T.; He, M.; Liu, R.J.; et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci. Immunol. 2017, 2, eaan4631. [Google Scholar] [CrossRef] [Green Version]
- Bueno-Martinez, E.; Lara-Almunia, M.; Rodriguez-Arias, C.; Otero-Rodriguez, A.; Garfias-Arjona, S.; Gonzalez-Sarmiento, R. Polymorphisms in autophagy genes are genetic susceptibility factors in glioblastoma development. BMC Cancer 2022, 22, 146. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Chen, Y.; Wei, X.; Wu, D.; Min, Z.; Quan, Y. Upregulated NTF4 in colorectal cancer promotes tumor development via regulating autophagy. Int. J. Oncol. 2020, 56, 1442–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.; Feng, L.; Bai, L.; Cui, H. NUCKS promotes cell proliferation and suppresses autophagy through the mTOR-Beclin1 pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2020, 39, 194. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gioia, M.; Spreafico, R.; Springstead, J.R.; Mendelson, M.M.; Joehanes, R.; Levy, D.; Zanoni, I. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat. Immunol. 2020, 21, 42–53. [Google Scholar] [CrossRef]
- Seok, J.K.; Hong, E.H.; Yang, G.; Lee, H.E.; Kim, S.E.; Liu, K.H.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy. Cells 2021, 10, 558. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Hu, J.; Wang, S.; Liu, Y.; Yang, M.; Zhang, J.; Tang, B. Inhibition of MGMT-mediated autophagy suppression decreases cisplatin chemosensitivity in gastric cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 125, 109896. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Easton, D.F.; Ford, D.; Bishop, D.T. Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1995, 56, 265–271. [Google Scholar]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Tekedereli, I.; Alpay, S.N.; Akar, U.; Yuca, E.; Ayugo-Rodriguez, C.; Han, H.D.; Sood, A.K.; Lopez-Berestein, G.; Ozpolat, B. Therapeutic Silencing of Bcl-2 by Systemically Administered siRNA Nanotherapeutics Inhibits Tumor Growth by Autophagy and Apoptosis and Enhances the Efficacy of Chemotherapy in Orthotopic Xenograft Models of ER (-) and ER (+) Breast Cancer. Mol. Nucleic Acids 2013, 2, e121. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Xiaofei, E.; Ni, D.; Pirooz, S.D.; Lee, J.Y.; Lee, D.; Zhao, Z.; Lee, S.; Lee, H.; Ku, B.; et al. Downregulation of autophagy by Bcl-2 promotes MCF7 breast cancer cell growth independent of its inhibition of apoptosis. Cell Death Differ. 2011, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. BCR 2013, 15, R109. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.H.; Kutty, M.K. Review of stem cell deregulation and breast cancer: An emerging hypothesis. Indian J. Pathol. Microbiol. 2012, 55, 147–153. [Google Scholar] [CrossRef]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.G.; Penfornis, P.; Oskowitz, A.Z.; Boonjindasup, A.G.; Cai, D.Z.; Dhule, S.S.; Rowan, B.G.; Kelekar, A.; Krause, D.S.; Pochampally, R.R. Activation of autophagy in mesenchymal stem cells provides tumor stromal support. Carcinogenesis 2011, 32, 964–972. [Google Scholar] [CrossRef]
- Kongara, S.; Kravchuk, O.; Teplova, I.; Lozy, F.; Schulte, J.; Moore, D.; Barnard, N.; Neumann, C.A.; White, E.; Karantza, V. Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol. Cancer Res. MCR 2010, 8, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, X.; Li, H.; Chen, Z.; Yao, X.; Jin, J.; Ma, X. TRPC5-induced autophagy promotes drug resistance in breast carcinoma via CaMKKbeta/AMPKalpha/mTOR pathway. Sci. Rep. 2017, 7, 3158. [Google Scholar] [CrossRef]
- Duan, L.; Motchoulski, N.; Danzer, B.; Davidovich, I.; Shariat-Madar, Z.; Levenson, V.V. Prolylcarboxypeptidase regulates proliferation, autophagy, and resistance to 4-hydroxytamoxifen-induced cytotoxicity in estrogen receptor-positive breast cancer cells. J. Biol. Chem. 2011, 286, 2864–2876. [Google Scholar] [CrossRef] [Green Version]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.; Rawson, J.; Smith, S.B.; Barrett, J.T.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 2008, 7, 2977–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Urruticoechea, A.; Martin-Castillo, B.; Menendez, J.A. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: Utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget 2012, 3, 1600–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Li, X.; Feng, J.; Chang, Y.; Wang, Z.; Wen, A. Autophagy facilitates the Lapatinib resistance of HER2 positive breast cancer cells. Med. Hypotheses 2011, 77, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.K.W.; Zeng, W.; Chen, J.; Yao, X.J.; Leung, E.L.H.; Wang, Q.Q.; Chiu, P.; Ko, B.C.B.; Law, B.Y.K. Tetrandrine, an Activator of Autophagy, Induces Autophagic Cell Death via PKC-alpha Inhibition and mTOR-Dependent Mechanisms. Front. Pharmacol. 2017, 8, 351. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin-induced autophagy leads to the apoptosis in breast cancer stem cells: Molecular mechanisms. Mol. Cancer 2013, 12, 171. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Zhang, X.; Qi, T.; Cheng, H.; Kong, Q.; Liu, L.; Cao, X.; Ding, Z. Alpha-lipoic acid inhibits lung cancer growth via mTOR-mediated autophagy inhibition. FEBS Open Bio. 2020, 10, 607–618. [Google Scholar] [CrossRef]
- Luo, J.; Yao, Y.; Ji, S.; Sun, Q.; Xu, Y.; Liu, K.; Diao, Q.; Qiang, Y.; Shen, Y. PITX2 enhances progression of lung adenocarcinoma by transcriptionally regulating WNT3A and activating Wnt/beta-catenin signaling pathway. Cancer Cell Int. 2019, 19, 96. [Google Scholar] [CrossRef]
- Wang, C.C.; Lin, S.Y.; Huang, Y.H.; Hsieh, C.H.; Chang, H.H.; Chen, H.Y.; Weng, C.W.; Chang, G.C.; Yu, S.L.; Chen, J.J. Paired-like homeodomain 2B contributes to tumour progression and anti-autophagy in human lung cancer. Am. J. Cancer Res. 2021, 11, 4900–4918. [Google Scholar] [PubMed]
- Baumgartner, U.; Berger, F.; Hashemi Gheinani, A.; Burgener, S.S.; Monastyrskaya, K.; Vassella, E. miR-19b enhances proliferation and apoptosis resistance via the EGFR signaling pathway by targeting PP2A and BIM in non-small cell lung cancer. Mol. Cancer 2018, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Feng, Y.; Qian, R. Up-regulation of microRNA miR-101-3p enhances sensitivity to cisplatin via regulation of small interfering RNA (siRNA) Anti-human AGT4D and autophagy in non-small-cell lung carcinoma (NSCLC). Bioengineered 2021, 12, 8435–8446. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.G.; Kim, S.Y.; Kim, H.R.; Kim, H.; Kim, C.H. Radiation Induces Autophagy via Histone H4 Lysine 20 Trimethylation in Non-small Cell Lung Cancer Cells. Anticancer Res. 2020, 40, 2537–2548. [Google Scholar] [CrossRef]
- Li, Y.Y.; Lam, S.K.; Zheng, C.Y.; Ho, J.C. The Effect of Tumor Microenvironment on Autophagy and Sensitivity to Targeted Therapy in EGFR-Mutated Lung Adenocarcinoma. J. Cancer 2015, 6, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Wadowski, B.; De Rienzo, A.; Bueno, R. The Molecular Basis of Malignant Pleural Mesothelioma. Thorac. Surg. Clin. 2020, 30, 383–393. [Google Scholar] [CrossRef]
- Friedberg, J.S.; Culligan, M.J.; Tsao, A.S.; Rusch, V.; Sepesi, B.; Pass, H.I.; Bueno, R.; Burt, B.; Sugarbaker, D.J.; de Perrot, M.; et al. A Proposed System Toward Standardizing Surgical-Based Treatments for Malignant Pleural Mesothelioma, From the Joint National Cancer Institute-International Association for the Study of Lung Cancer-Mesothelioma Applied Research Foundation Taskforce. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017, 24, 1694–1704. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Giorgi, C.; Galluzzi, L.; Pinton, P. Ca2+ Fluxes and Cancer. Mol. Cell 2020, 78, 1055–1069. [Google Scholar] [CrossRef]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.H.; Rangarajan, A. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 2009, 83, 8565–8574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Okimoto, G.; Jube, S.; Napolitano, A.; Pass, H.I.; Laczko, R.; Demay, R.M.; Khan, G.; Tiirikainen, M.; Rinaudo, C.; et al. Continuous exposure to chrysotile asbestos can cause transformation of human mesothelial cells via HMGB1 and TNF-alpha signaling. Am. J. Pathol. 2013, 183, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, A.; Antoine, D.J.; Pellegrini, L.; Baumann, F.; Pagano, I.; Pastorino, S.; Goparaju, C.M.; Prokrym, K.; Canino, C.; Pass, H.I.; et al. HMGB1 and Its Hyperacetylated Isoform are Sensitive and Specific Serum Biomarkers to Detect Asbestos Exposure and to Identify Mesothelioma Patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Patergnani, S.; Giorgi, C.; Suarez, J.; Goto, K.; Bononi, A.; Tanji, M.; Novelli, F.; Pastorino, S.; Xu, R.; et al. Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy. Proc. Natl. Acad. Sci. USA 2020, 117, 25543–25552. [Google Scholar] [CrossRef]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. 2015, 6, e1757. [Google Scholar] [CrossRef] [Green Version]
- Barbone, D.; Follo, C.; Echeverry, N.; Gerbaudo, V.H.; Klabatsa, A.; Bueno, R.; Felley-Bosco, E.; Broaddus, V.C. Autophagy Correlates with the Therapeutic Responsiveness of Malignant Pleural Mesothelioma in 3D Models. PLoS ONE 2015, 10, e0134825. [Google Scholar] [CrossRef] [Green Version]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol. Carcinog. 2018, 57, 319–332. [Google Scholar] [CrossRef]
- Xu, D.; Liang, S.Q.; Yang, Z.; Yang, H.; Bruggmann, R.; Oberhaensli, S.; Berezowska, S.; Marti, T.M.; Hall, S.R.R.; Dorn, P.; et al. Malignant pleural mesothelioma co-opts BCL-XL and autophagy to escape apoptosis. Cell Death Dis. 2021, 12, 406. [Google Scholar] [CrossRef]
- Kenner, B.J.; Chari, S.T.; Maitra, A.; Srivastava, S.; Cleeter, D.F.; Go, V.L.; Rothschild, L.J.; Goldberg, A.E. Early Detection of Pancreatic Cancer-a Defined Future Using Lessons from Other Cancers: A White Paper. Pancreas 2016, 45, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, B.; Mohd-Naim, N.; Maximiano, F.; Frasa, M.A.; McCormack, J.; Finelli, M.; Thoresen, S.B.; Perdios, L.; Daigaku, R.; Francis, R.E.; et al. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev. Cell 2013, 25, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totaro, A.; Zhuang, Q.; Panciera, T.; Battilana, G.; Azzolin, L.; Brumana, G.; Gandin, A.; Brusatin, G.; Cordenonsi, M.; Piccolo, S. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc. Natl. Acad. Sci. USA 2019, 116, 17848–17857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Yang, M.C.; Wang, H.C.; Hou, Y.C.; Tung, H.L.; Chiu, T.J.; Shan, Y.S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1alpha promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [Green Version]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Lam, S.K.; Mak, J.C.; Zheng, C.Y.; Ho, J.C. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Hernandez, M.; Arias, A.; Martinez-Garcia, D.; Perez-Tomas, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Sulkshane, P.; Teni, T. BH3 mimetic Obatoclax (GX15-070) mediates mitochondrial stress predominantly via MCL-1 inhibition and induces autophagy-dependent necroptosis in human oral cancer cells. Oncotarget 2017, 8, 60060–60079. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [Green Version]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, X.; Guo, H.; Zhang, B.; Zhang, X.B.; Shi, Z.J.; Yu, L. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy 2013, 9, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anti-Cancer Drugs 2017, 28, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.F.; Tsai, K.L.; Hsu, C.J.; Tsai, W.L.; Cheng, J.S.; Chang, H.W.; Shiau, C.W.; Goan, Y.G.; Tseng, H.H.; Wu, C.H.; et al. Drug Repurposing Screening Identifies Tioconazole as an ATG4 Inhibitor that Suppresses Autophagy and Sensitizes Cancer Cells to Chemotherapy. Theranostics 2018, 8, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Q.; Xie, W.J.; Pan, D.; Chen, H.; Zhang, L. Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumour Biol. 2016, 37, 653–659. [Google Scholar] [CrossRef]
- Yin, Y.C.; Lin, C.C.; Chen, T.T.; Chen, J.Y.; Tsai, H.J.; Wang, C.Y.; Chen, S.Y. Clozapine induces autophagic cell death in non-small cell lung cancer cells. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 35, 945–956. [Google Scholar] [CrossRef]
- Rossi, M.; Munarriz, E.R.; Bartesaghi, S.; Milanese, M.; Dinsdale, D.; Guerra-Martin, M.A.; Bampton, E.T.; Glynn, P.; Bonanno, G.; Knight, R.A.; et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J. Cell Sci. 2009, 122, 3330–3339. [Google Scholar] [CrossRef] [Green Version]
- Khing, T.M.; Po, W.W.; Sohn, U.D. Fluoxetine Enhances Anti-tumor Activity of Paclitaxel in Gastric Adenocarcinoma Cells by Triggering Apoptosis and Necroptosis. Anticancer Res. 2019, 39, 6155–6163. [Google Scholar] [CrossRef]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuto, M.; Mattera, R.; Sticca, J.I.; Rossi, P.; Cipriani, C.; Giganti, M.G.; Volpi, A.; Modesti, A.; Masuelli, L.; Bei, R. Effect of the BH3 Mimetic Polyphenol (-)-Gossypol (AT-101) on the in vitro and in vivo Growth of Malignant Mesothelioma. Front. Pharmacol. 2018, 9, 1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuto, M.; Mattera, R.; Masuelli, L.; Taffera, G.; Andracchio, O.; Tresoldi, I.; Lido, P.; Giganti, M.G.; Godos, J.; Modesti, A.; et al. (+/−)-Gossypol induces apoptosis and autophagy in head and neck carcinoma cell lines and inhibits the growth of transplanted salivary gland cancer cells in BALB/c mice. Int. J. Food Sci. Nutr. 2017, 68, 298–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Lindmo, K.; Stenmark, H. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 2006, 119, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS ONE 2013, 8, e55096. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
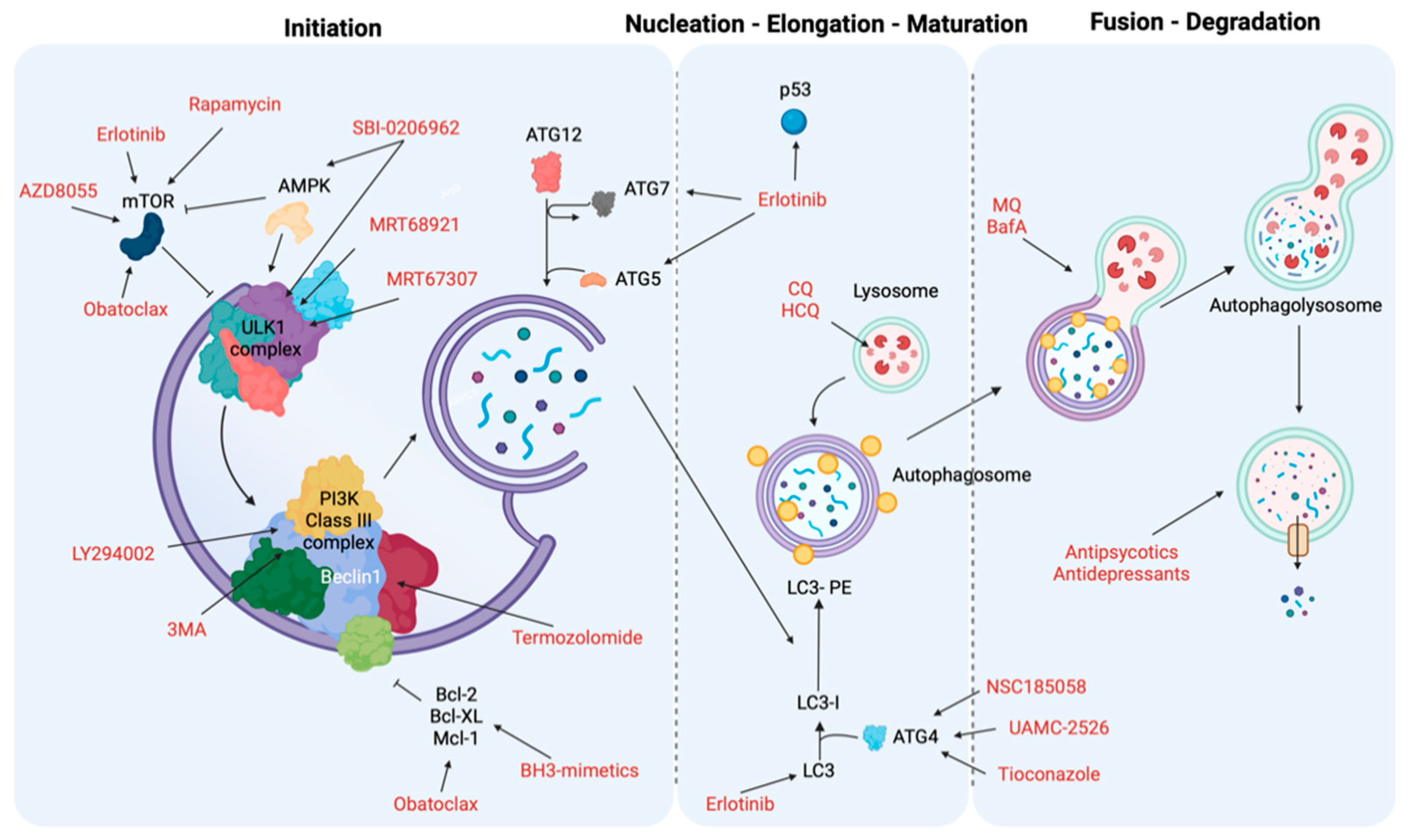
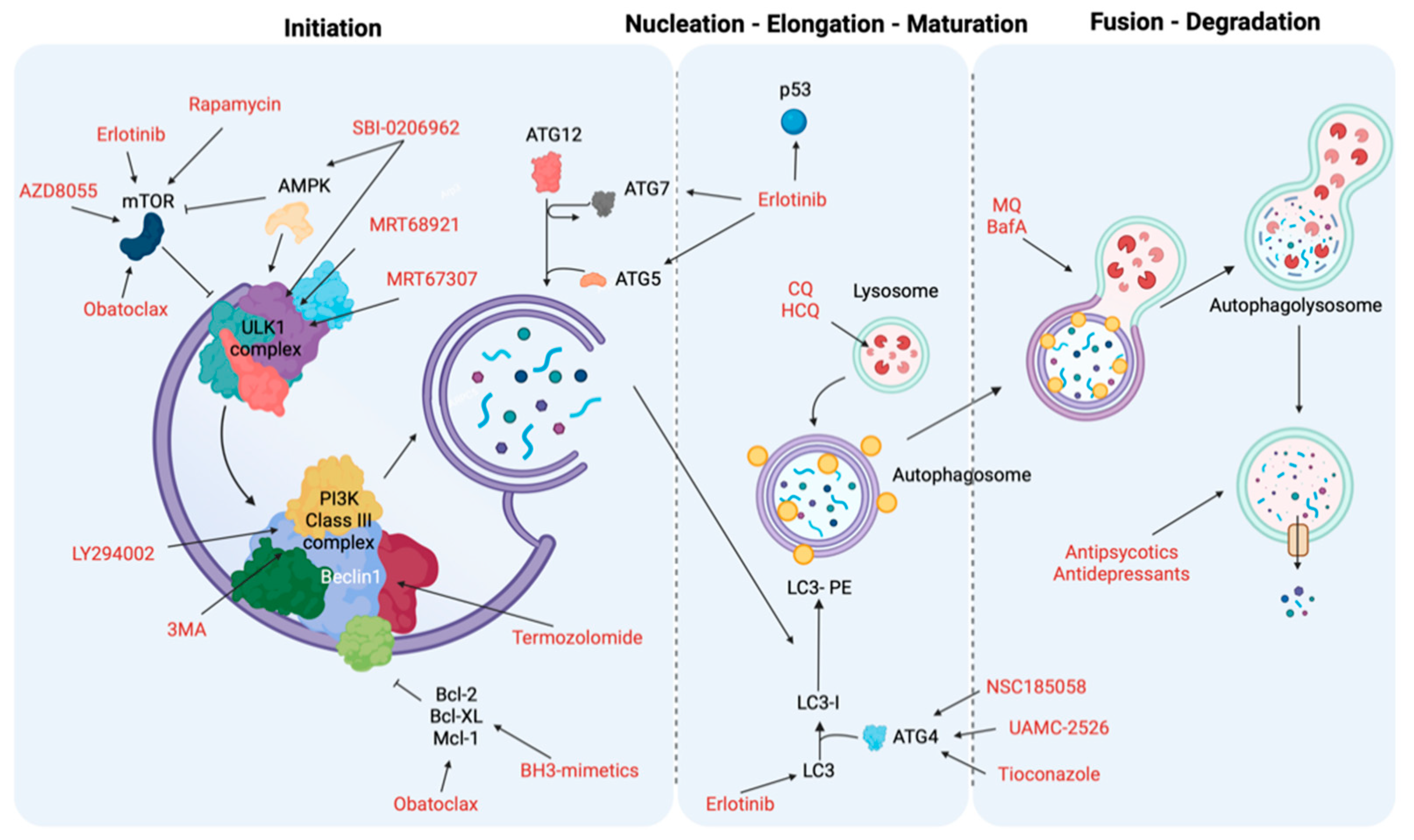
| Autophagy Phases | Treatment | Target | Cancer Type | Ref. |
|---|---|---|---|---|
| Initiation and nucleation phases | Erlotinib | TK domain, mTOR and p53 | NSCLC | [231] |
| Sorafenib | AKT | Hepatocellular carcinoma (HCC) | [232] | |
| Rapamycin | mTORC1 | Several | [233,234] | |
| AZD8055 | mTOR | Several | [235] | |
| BH3 mimetics | BH3-proteins (Bcl-2, Mcl-1) and mTOR | Malignant mesothelioma, colon cancer cells, oral squamous cell carcinoma and acute lymphoblastic leukemia cells | [236,237] | |
| MRT68921 and MRT67307 | ULK1 and ULK2 | Several | [238] | |
| SBI-0206965 | ULK1 and AMPK | Renal cell carcinoma and neuroblastoma | [238] | |
| 3-methyladenine (3MA) | PI3K complex | Several | [239,240] | |
| LY294002 | PI3K complex | Melanoma | [241] | |
| Elongation and closure | NSC185058 | ATG4 | Subcutaneous osteosarcoma | [242] |
| UAMC-2526 | ATG proteins | Murine colorectal cancer | [243] | |
| Tioconazole | ATG proteins | Several | [244] | |
| Vesicle degradation | Chloroquine (CQ) and hydroxychloroquine (HCQ) | Lysosomes | Colon cancer, melanoma cells and breast cancer | [245,246] |
| Mefloquine (MQ) and Bafilomycin A (BafA) | Autophagosome-lysosome | Chronic myeloid leukemia, prostate cancer and breast cancers | [247] | |
| Antipsychotics or Antidepressants | Lysosomes | NSCLC, gastric cancer and breast cancer | [248,249,250,251] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitto, V.A.M.; Bianchin, S.; Zolondick, A.A.; Pellielo, G.; Rimessi, A.; Chianese, D.; Yang, H.; Carbone, M.; Pinton, P.; Giorgi, C.; et al. Molecular Mechanisms of Autophagy in Cancer Development, Progression, and Therapy. Biomedicines 2022, 10, 1596. https://doi.org/10.3390/biomedicines10071596
Vitto VAM, Bianchin S, Zolondick AA, Pellielo G, Rimessi A, Chianese D, Yang H, Carbone M, Pinton P, Giorgi C, et al. Molecular Mechanisms of Autophagy in Cancer Development, Progression, and Therapy. Biomedicines. 2022; 10(7):1596. https://doi.org/10.3390/biomedicines10071596
Chicago/Turabian StyleVitto, Veronica Angela Maria, Silvia Bianchin, Alicia Ann Zolondick, Giulia Pellielo, Alessandro Rimessi, Diego Chianese, Haining Yang, Michele Carbone, Paolo Pinton, Carlotta Giorgi, and et al. 2022. "Molecular Mechanisms of Autophagy in Cancer Development, Progression, and Therapy" Biomedicines 10, no. 7: 1596. https://doi.org/10.3390/biomedicines10071596
APA StyleVitto, V. A. M., Bianchin, S., Zolondick, A. A., Pellielo, G., Rimessi, A., Chianese, D., Yang, H., Carbone, M., Pinton, P., Giorgi, C., & Patergnani, S. (2022). Molecular Mechanisms of Autophagy in Cancer Development, Progression, and Therapy. Biomedicines, 10(7), 1596. https://doi.org/10.3390/biomedicines10071596