
Mapping the Serum Proteome of COVID-19 Patients; Guidance for Severity Assessment

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
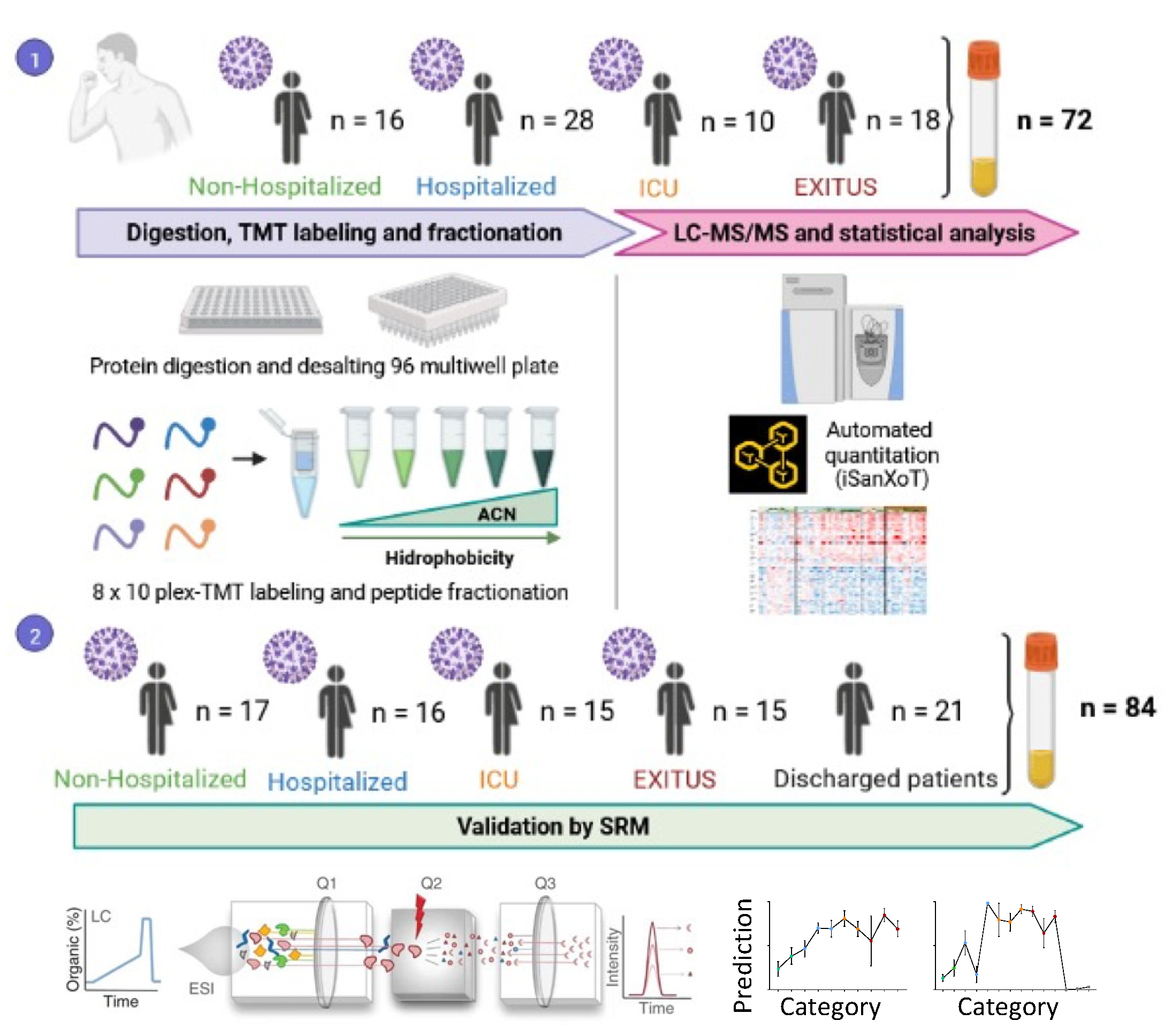
2.1. Human Samples
2.2. Sample Preparation
2.3. Multiplexed Isobaric TMT Labeling
2.4. Peptide Fractionation
2.5. LC-MS Analysis
2.6. Protein Identification
2.7. Protein Quantification and Statistical Analysis
2.8. Peptide Synthesis
2.9. SRM Analysis
2.10. Statistical Analysis
3. Results
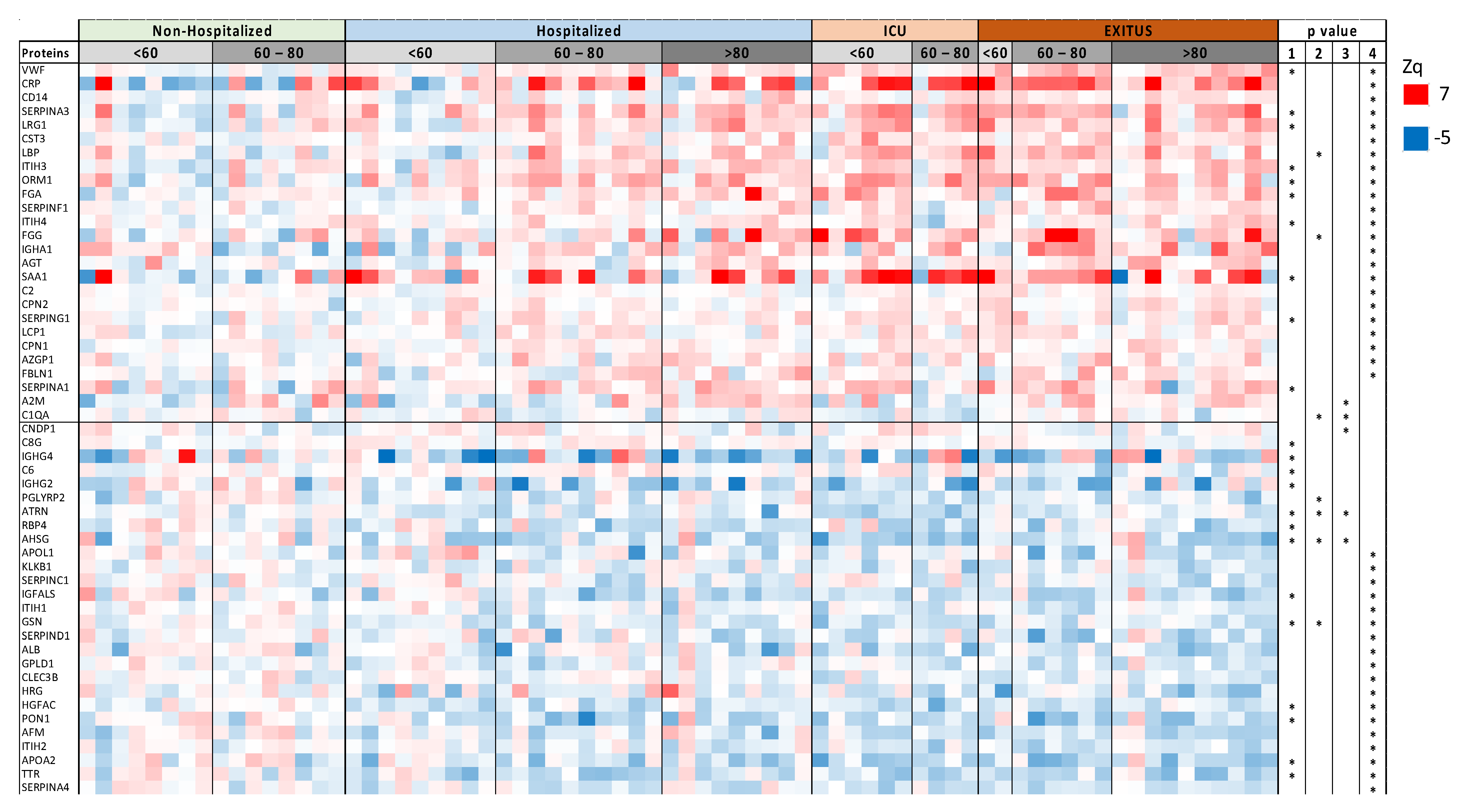
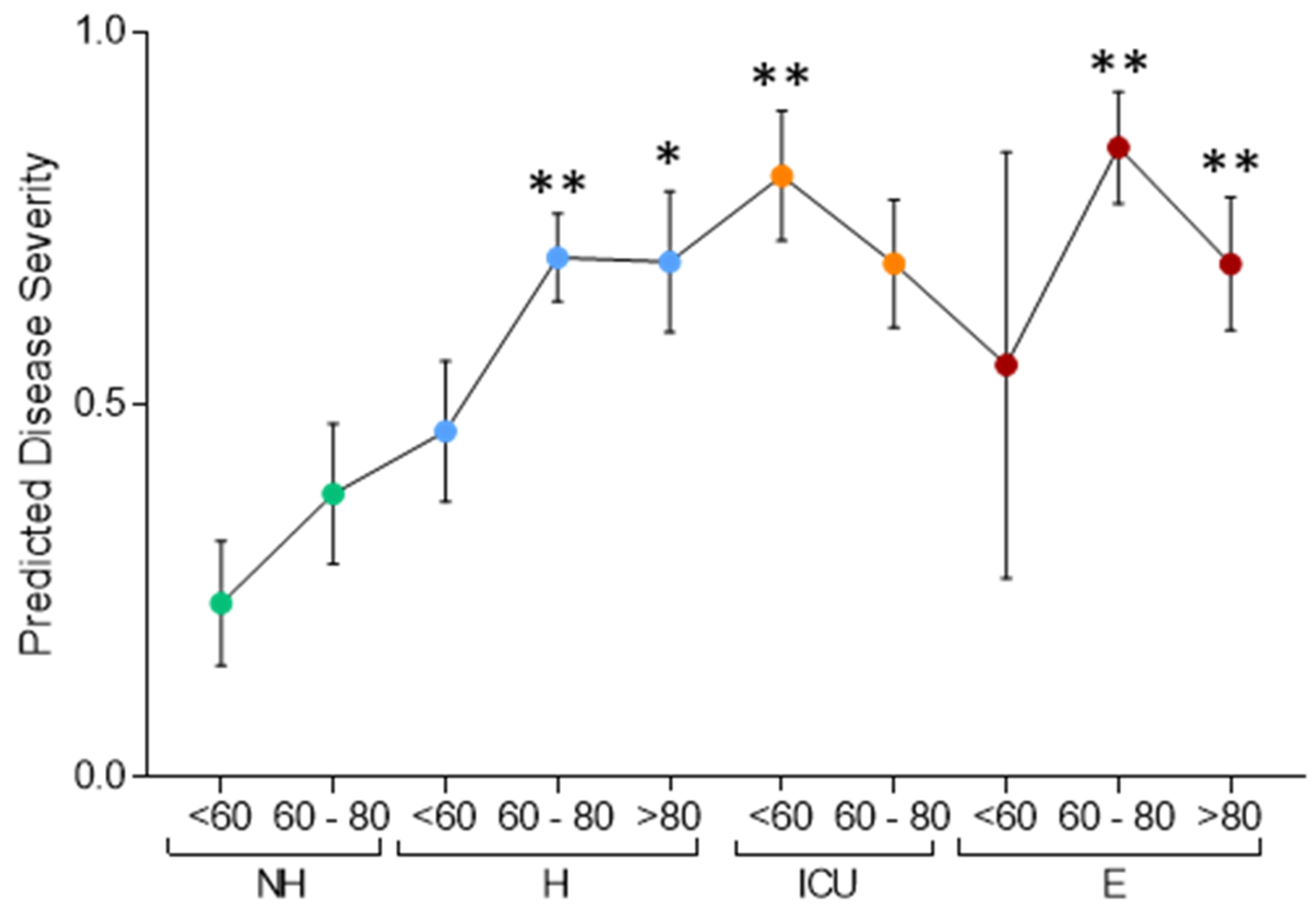
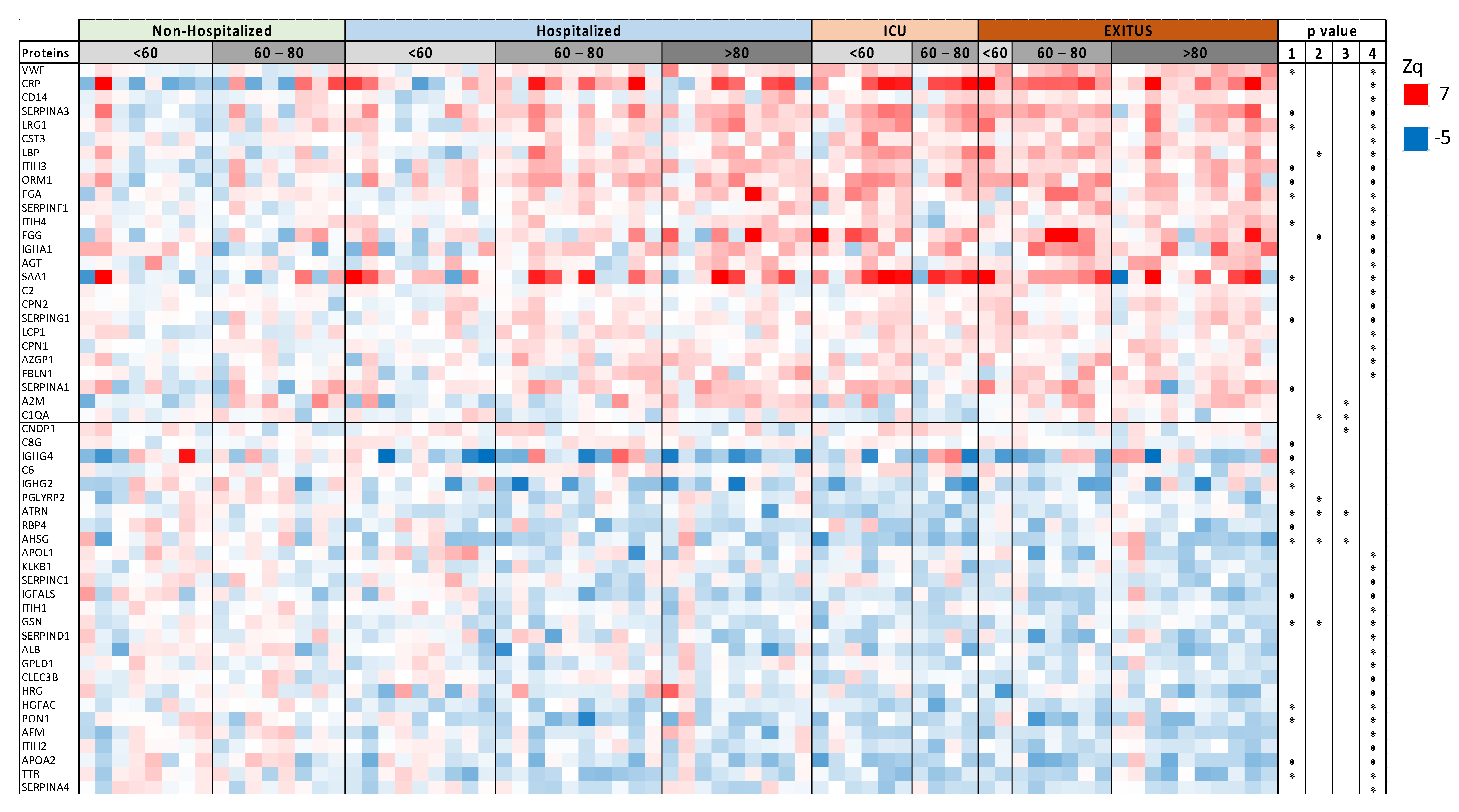
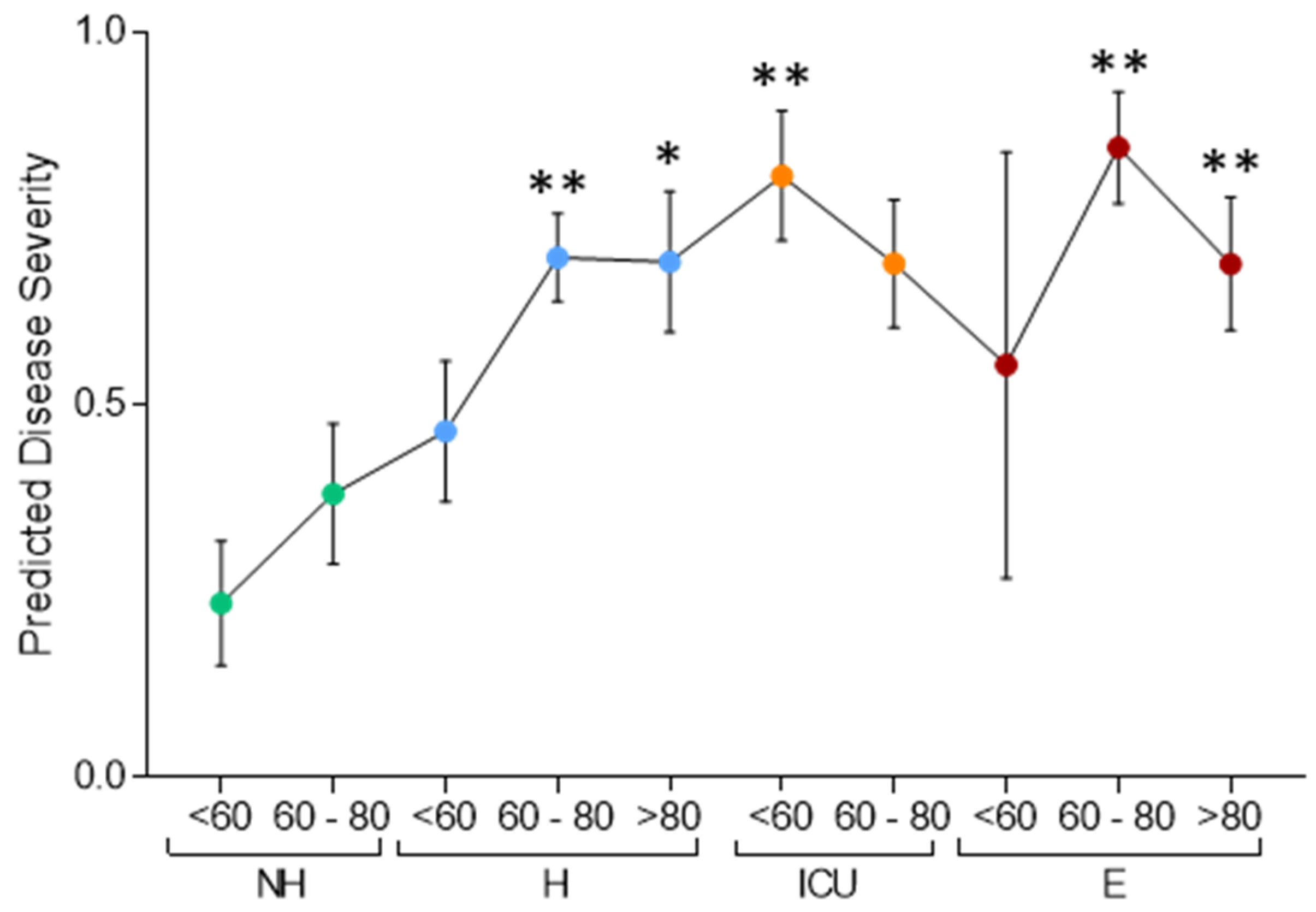
3.1. Serum Proteomic Analysis of the Discovery Cohort
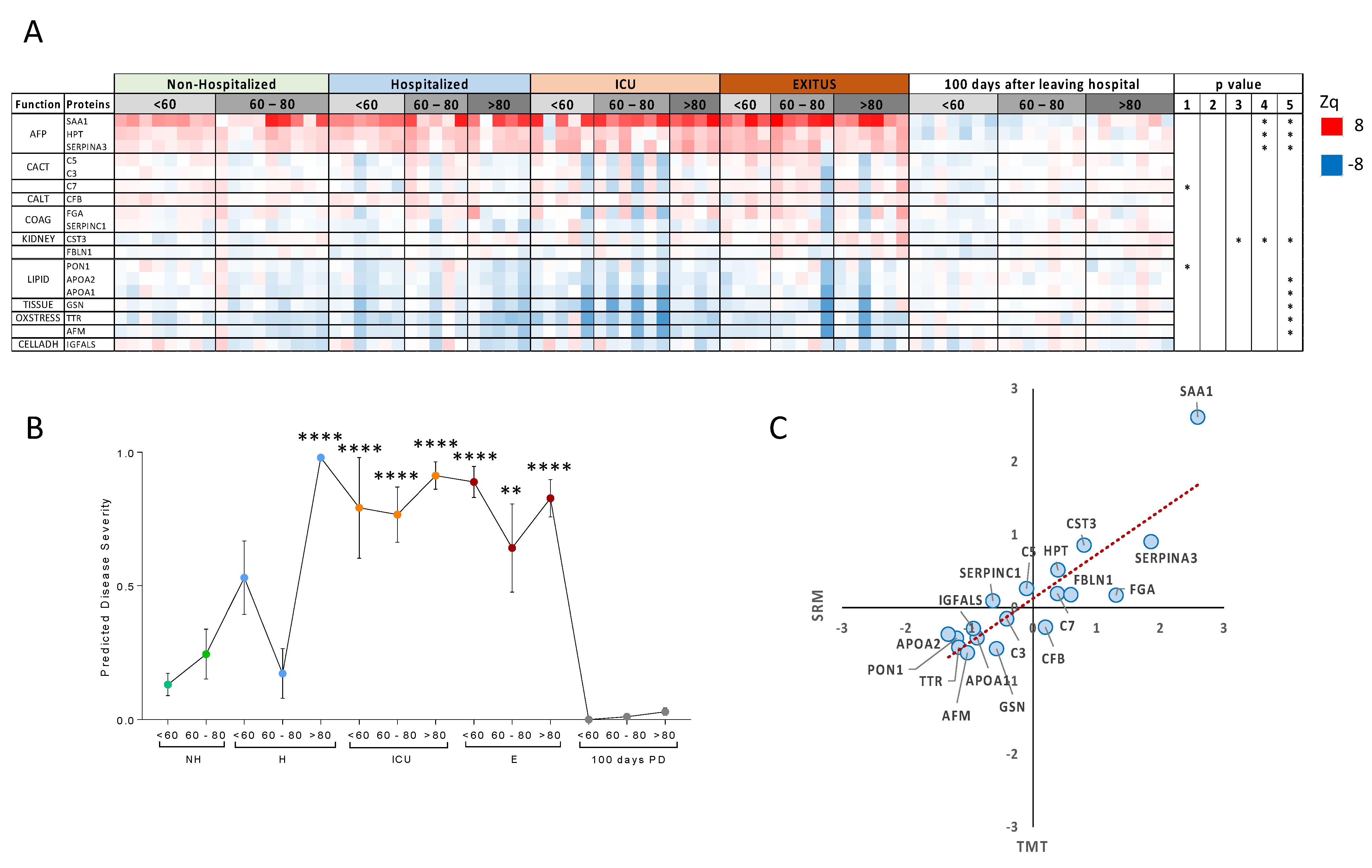
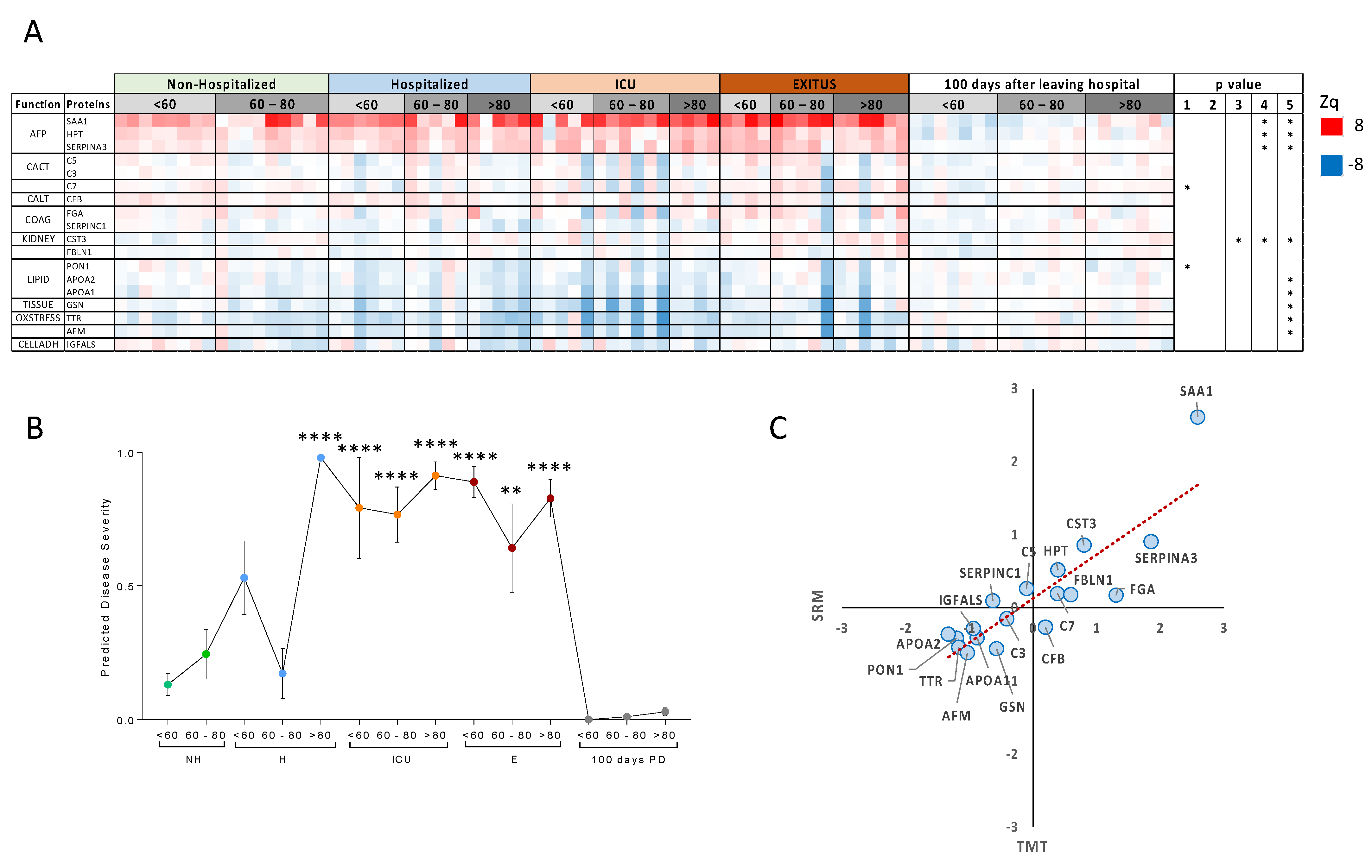
3.2. Targeted Analysis in the Validation Cohort
3.3. Comparison with Other Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e0012720. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and Coronavirus Disease-2019 (COVID-19): The Epidemic and the Challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Similarities and Dissimilarities of COVID-19 and Other Coronavirus Diseases. Annu. Rev. Microbiol. 2021, 75, 19–47. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, X.; Qiu, Y.; Song, Y.; Feng, F.; Feng, J.; Song, Q.; Jia, Q.; Wang, J. Clinical Characteristics of 82 Cases of Death from COVID-19. PLoS ONE 2020, 15, e0235458. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical Course and Outcomes of Critically Ill Patients with SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA J. Am. Med. Assoc. 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Völlmy, F.; van den Toorn, H.; Chiozzi, R.Z.; Zucchetti, O.; Papi, A.; Volta, C.A.; Marracino, L.; Sega, F.V.D.; Fortini, F.; Demichev, V.; et al. A Serum Proteome Signature to Predict Mortality in Severe COVID-19 Patients. Life Sci. Alliance 2021, 4, e202101099. [Google Scholar] [CrossRef]
- Demichev, V.; Tober-Lau, P.; Lemke, O.; Nazarenko, T.; Thibeault, C.; Whitwell, H.; Röhl, A.; Freiwald, A.; Szyrwiel, L.; Ludwig, D.; et al. A Time-Resolved Proteomic and Prognostic Map of COVID-19. Cell Syst. 2021, 12, 780–794.e7. [Google Scholar] [CrossRef]
- Geyer, P.E.; Arend, F.M.; Doll, S.; Louiset, M.; Virreira Winter, S.; Müller-Reif, J.B.; Torun, F.M.; Weigand, M.; Eichhorn, P.; Bruegel, M.; et al. High-resolution Serum Proteome Trajectories in COVID-19 Reveal Patient-specific Seroconversion. EMBO Mol. Med. 2021, 13, e14167. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Baxter, R.; Araya, P.; Jordan, K.R.; Russell, S.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; et al. Seroconversion Stages COVID19 into Distinct Pathophysiological States. eLife 2021, 10, e65508. [Google Scholar] [CrossRef]
- Suvarna, K.; Biswas, D.; Pai, M.G.J.; Acharjee, A.; Bankar, R.; Palanivel, V.; Salkar, A.; Verma, A.; Mukherjee, A.; Choudhury, M.; et al. Proteomics and Machine Learning Approaches Reveal a Set of Prognostic Markers for COVID-19 Severity with Drug Repurposing Potential. Front. Physiol. 2021, 12, 652799. [Google Scholar] [CrossRef]
- Yu, Y.T.C.; Chien, S.C.; Chen, I.Y.; Lai, C.T.; Tsay, Y.G.; Chang, S.C.; Chang, M.F. Surface Vimentin Is Critical for the Cell Entry of SARS-CoV. J. Biomed. Sci. 2016, 23, 14. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738.e13. [Google Scholar] [CrossRef]
- Liu, C.; Mendonça, L.; Yang, Y.; Gao, Y.; Shen, C.; Liu, J.; Ni, T.; Ju, B.; Liu, C.; Tang, X.; et al. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure 2020, 28, 1218–1224.e4. [Google Scholar] [CrossRef]
- Srzentić, K.; Fornelli, L.; Tsybin, Y.O.; Loo, J.A.; Seckler, H.; Agar, J.N.; Anderson, L.C.; Bai, D.L.; Beck, A.; Brodbelt, J.S.; et al. Interlaboratory Study for Characterizing Monoclonal Antibodies by Top-Down and Middle-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 1783–1802. [Google Scholar] [CrossRef]
- Resemann, A.; Liu-Shin, L.; Tremintin, G.; Malhotra, A.; Fung, A.; Wang, F.; Ratnaswamy, G.; Suckau, D. Rapid, Automated Characterization of Disulfide Bond Scrambling and IgG2 Isoform Determination. mAbs 2018, 10, 1200–1213. [Google Scholar] [CrossRef]
- Martínez-Bartolomé, S.; Navarro, P.; Martín-Maroto, F.; López-Ferrer, D.; Ramos-Fernández, A.; Villar, M.; García-Ruiz, J.P.; Vázquez, J. Properties of Average Score Distributions of SEQUEST: The Probability Ratio Method. Mol. Cell. Proteom. 2008, 7, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Navarro, P.; Vazquez, J. A Refined Method to Calculate False Discovery Rates for Peptide Identification Using Decoy Databases. J. Proteome Res. 2009, 8, 1792–1796. [Google Scholar] [CrossRef]
- Bonzon-Kulichenko, E.; Garcia-Marques, F.; Trevisan-Herraz, M.; Vázquez, J. Revisiting Peptide Identification by High-Accuracy Mass Spectrometry: Problems Associated with the Use of Narrow Mass Precursor Windows. J. Proteome Res. 2015, 14, 700–710. [Google Scholar] [CrossRef]
- Navarro, P.; Trevisan-Herraz, M.; Bonzon-Kulichenko, E.; Núñez, E.; Martínez-Acedo, P.; Pérez-Hernández, D.; Jorge, I.; Mesa, R.; Calvo, E.; Carrascal, M.; et al. General Statistical Framework for Quantitative Proteomics by Stable Isotope Labeling. J. Proteome Res. 2014, 13, 1234–1247. [Google Scholar] [CrossRef]
- Jorge, I.; Navarro, P.; Martínez-Acedo, P.; Núñez, E.; Serrano, H.; Alfranca, A.; Redondo, J.M.; Vázquez, J. Statistical Model to Analyze Quantitative Proteomics Data Obtained by 18O/16O Labeling and Linear Ion Trap Mass Spectrometry: Application to the Study of Vascular Endothelial Growth Factor-Induced Angiogenesis in Endothelial Cells. Mol. Cell. Proteom. 2009, 8, 1130–1149. [Google Scholar] [CrossRef] [Green Version]
- García-Marqués, F.; Trevisan-Herraz, M.; Martínez-Martínez, S.; Camafeita, E.; Jorge, I.; Lopez, J.A.; Méndez-Barbero, N.; Méndez-Ferrer, S.; del Pozo, M.A.; Ibáñez, B.; et al. A Novel Systems-Biology Algorithm for the Analysis of Coordinated Protein Responses Using Quantitative Proteomics. Mol. Cell. Proteom. 2016, 15, 1740–1760. [Google Scholar] [CrossRef] [Green Version]
- Trevisan-Herraz, M.; Bagwan, N.; García-Marqués, F.; Rodriguez, J.M.; Jorge, I.; Ezkurdia, I.; Bonzon-Kulichenko, E.; Vázquez, J. SanXoT: A Modular and Versatile Package for the Quantitative Analysis of High-Throughput Proteomics Experiments. Bioinformatics 2019, 35, 1594–1596. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Núñez, E.; Fuster, V.; Gómez-Serrano, M.; Valdivielso, J.M.; Fernández-Alvira, J.M.; Martínez-López, D.; Rodríguez, J.M.; Bonzon-Kulichenko, E.; Calvo, E.; Alfayate, A.; et al. Unbiased Plasma Proteomics Discovery of Biomarkers for Improved Detection of Subclinical Atherosclerosis. eBioMedicine 2022, 76, 103874. [Google Scholar] [CrossRef]
- Corbacho-Alonso, N.; Baldán-Martín, M.; López, J.A.; Rodríguez-Sánchez, E.; Martínez, P.J.; Mourino-Alvarez, L.; Sastre-Oliva, T.; Cabrera, M.; Calvo, E.; Padial, L.R.; et al. Cardiovascular Risk Stratification Based on Oxidative Stress for Early Detection of Pathology. Antioxid. Redox Signal. 2021, 35, 602–617. [Google Scholar] [CrossRef]
- Corbacho-Alonso, N.; Baldán-Martín, M.; López, J.A.; Rodríguez Sánchez, E.; Martínez, P.J.; Mourino-Alvarez, L.; Martin-Rojas, T.; Sastre-Oliva, T.; Madruga, F.; Vázquez, J.; et al. Novel Molecular Plasma Signatures on Cardiovascular Disease Can Stratify Patients throughout Life. J. Proteom. 2020, 222, 103816. [Google Scholar] [CrossRef]
- Calvo, E.; Corbacho-Alonso, N.; Sastre-Oliva, T.; Nuñez, E.; Baena-Galan, P.; Hernandez-Fernandez, G.; Rodriguez-Cola, M.; Jimenez-Velasco, I.; Corrales, F.J.; Gambarrutta-Malfati, C.; et al. Why Does COVID-19 Affect Patients with Spinal Cord Injury Milder? A Case-Control Study: Results from Two Observational Cohorts. J. Pers. Med. 2020, 10, 182. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Alonso, A.; Toral, M.; Alfayate, A.; Ruiz-Rodríguez, M.J.; Bonzón-Kulichenko, E.; Teixido-Tura, G.; Martínez-Martínez, S.; Méndez-Olivares, M.J.; López-Maderuelo, D.; González-Valdés, I.; et al. Aortic Disease in Marfan Syndrome Is Caused by Overactivation of SGC-PRKG Signaling by NO. Nat. Commun. 2021, 12, 2628. [Google Scholar] [CrossRef] [PubMed]
- Santos-Lozano, A.; Fiuza-Luces, C.; Fernández-Moreno, D.; Llavero, F.; Arenas, J.; López, J.A.; Vázquez, J.; Escribano-Subías, P.; Zugaza, J.L.; Lucia, A. Exercise Benefits in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2019, 73, 2906–2907. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Thomas, T.; Dzieciatkowska, M.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hod, E.A.; Spitalnik, S.L.; Hansen, K.C. Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. J. Proteome Res. 2020, 19, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Messner, C.B.; Demichev, V.; Bloomfield, N.; Yu, J.S.L.; White, M.; Kreidl, M.; Egger, A.S.; Freiwald, A.; Ivosev, G.; Wasim, F.; et al. Ultra-Fast Proteomics with Scanning SWATH. Nat. Biotechnol. 2021, 39, 846–854. [Google Scholar] [CrossRef]
- Zhong, W.; Altay, O.; Arif, M.; Edfors, F.; Doganay, L.; Mardinoglu, A.; Uhlen, M.; Fagerberg, L. Next Generation Plasma Proteome Profiling of COVID-19 Patients with Mild to Moderate Symptoms. eBioMedicine 2021, 74, 103723. [Google Scholar] [CrossRef]
- Li, Y.; Schneider, A.M.; Mehta, A.; Sade-Feldman, M.; Kays, K.R.; Gentili, M.; Charland, N.C.; Gonye, A.L.K.; Gushterova, I.; Khanna, H.K.; et al. SARS-CoV-2 Viremia Is Associated with Distinct Proteomic Pathways and Predicts COVID-19 Outcomes. J. Clin. Investig. 2021, 131, e148635. [Google Scholar] [CrossRef]
- Al-Nesf, M.A.Y.; Abdesselem, H.B.; Bensmail, I.; Ibrahim, S.; Saeed, W.A.H.; Mohammed, S.S.I.; Razok, A.; Alhussain, H.; Aly, R.M.A.; Al-Maslamani, M.; et al. Prognostic Tools and Candidate Drugs Based on Plasma Proteomics of Patients with Severe COVID-19 Complications. Nat. Commun. 2022, 13, 946. [Google Scholar] [CrossRef]
- Bauer, W.; Weber, M.; Diehl-Wiesenecker, E.; Galtung, N.; Prpic, M.; Somasundaram, R.; Tauber, R.; Schwenk, J.M.; Micke, P.; Kappert, K. Plasma Proteome Fingerprints Reveal Distinctiveness and Clinical Outcome of SARS-CoV-2 Infection. Viruses 2021, 13, 2456. [Google Scholar] [CrossRef]
- Mohammed, Y.; Goodlett, D.R.; Cheng, M.P.; Vinh, D.C.; Lee, T.C.; McGeer, A.; Sweet, D.; Tran, K.; Lee, T.; Murthy, S.; et al. Longitudinal Plasma Proteomics Analysis Reveals Novel Candidate Biomarkers in Acute COVID-19. J. Proteome Res. 2022, 21, 975–992. [Google Scholar] [CrossRef] [PubMed]
- Calvet, J.; Berenguer-Llergo, A.; Gay, M.; Massanella, M.; Domingo, P.; Llop, M.; Sánchez-Jiménez, E.; Arévalo, M.; Carrillo, J.; Albiñana, N.; et al. Biomarker Candidates for Progression and Clinical Management of COVID-19 Associated Pneumonia at Time of Admission. Sci. Rep. 2022, 12, 640. [Google Scholar] [CrossRef] [PubMed]
- Ahern, D.J.; Ai, Z.; Ainsworth, M.; Allan, C.; Allcock, A.; Angus, B.; Ansari, M.A.; Arancibia-Cárcamo, C.v.; Aschenbrenner, D.; Attar, M.; et al. A Blood Atlas of COVID-19 Defines Hallmarks of Disease Severity and Specificity. Cell 2022, 185, 916–938.e58. [Google Scholar] [CrossRef]
- Park, J.; Kim, H.; Kim, S.Y.; Kim, Y.; Lee, J.S.; Dan, K.; Seong, M.W.; Han, D. In-Depth Blood Proteome Profiling Analysis Revealed Distinct Functional Characteristics of Plasma Proteins between Severe and Non-Severe COVID-19 Patients. Sci. Rep. 2020, 10, 22418. [Google Scholar] [CrossRef] [PubMed]
- Shu, T.; Ning, W.; Wu, D.; Xu, J.; Han, Q.; Huang, M.; Zou, X.; Yang, Q.; Yuan, Y.; Bie, Y.; et al. Plasma Proteomics Identify Biomarkers and Pathogenesis of COVID-19. Immunity 2020, 53, 1108–1122.e5. [Google Scholar] [CrossRef] [PubMed]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24.e4. [Google Scholar] [CrossRef] [PubMed]
- Coomes, E.A.; Haghbayan, H. Interleukin-6 in COVID-19: A Systematic Review and Meta-Analysis. Rev. Med. Virol. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- Lord, M.S.; Melrose, J.; Day, A.J.; Whitelock, J.M. The Inter-α-Trypsin Inhibitor Family: Versatile Molecules in Biology and Pathology. J. Histochem. Cytochem. 2020, 68, 907–927. [Google Scholar] [CrossRef]
- Nakajima, H.; Nakajima, K.; Serada, S.; Fujimoto, M.; Naka, T.; Sano, S. The Involvement of Leucine-Rich α-2 Glycoprotein in the Progression of Skin and Lung Fibrosis in Bleomycin-Induced Systemic Sclerosis Model. Mod. Rheumatol. 2021, 31, 1120–1128. [Google Scholar] [CrossRef]
- Lind, S.E.; Smith, D.B.; Janmey, P.A.; Stossel, T.P. Depression of Gelsolin Levels and Detection of Gelsolin-Actin Complexes in Plasma of Patients with Acute Lung Injury. Am. Rev. Respir. Dis. 1988, 138, 429–434. [Google Scholar] [CrossRef]
- Ito, H.; Kambe, H.; Kimura, Y.; Nakamura, H.; Hayashi, E.; Kishimoto, T.; Kishimoto, S.; Yamamoto, H. Depression of Plasma Gelsolin Level during Acute Liver Injury. Gastroenterology 1992, 102, 1686–1692. [Google Scholar] [CrossRef]
- Suhler, E.; Lin, W.; Yin, H.L.; Lee, W.M. Decreased Plasma Gelsolin Concentrations in Acute Liver Failure, Myocardial Infarction, Septic Shock, and Myonecrosis. Crit. Care Med. 1997, 25, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Osborn, T.M.; Verdrengh, M.; Stossel, T.P.; Bokarewa, M. Decreased Levels of the Gelsolin Plasma Isoform in Patients with Rheumatoid Arthritis. Arthritis Res. Ther. 2008, 10, R117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onopiuk, A.; Tokarzewicz, A.; Gorodkiewicz, E. Cystatin C: A Kidney Function Biomarker. In Advances in Clinical Chemistry; Academic Press Inc.: Cambridge, MA, USA, 2015; Volume 68, pp. 57–69. [Google Scholar]
- Brankovic, M.; Akkerhuis, K.M.; Buljubasic, N.; Cheng, J.M.; Oemrawsingh, R.M.; Garcia-Garcia, H.M.; Regar, E.; Serruys, P.W.; van Geuns, R.J.; Boersma, E.; et al. Plasma Cystatin C and Neutrophil Gelatinase-Associated Lipocalin in Relation to Coronary Atherosclerosis on Intravascular Ultrasound and Cardiovascular Outcome: Impact of Kidney Function (ATHEROREMO-IVUS Study). Atherosclerosis 2016, 254, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imig, J.D.; Ryan, M.J. Immune and Inflammatory Role in Renal Disease. Compr. Physiol. 2013, 3, 957–976. [Google Scholar] [CrossRef] [Green Version]
- Eddington, H.; Sinha, S.; Kalra, P.A. Vascular Calcification in Chronic Kidney Disease: A Clinical Review. J. Ren. Care 2009, 35, 45–50. [Google Scholar] [CrossRef]
- Ronco, C.; Reis, T.; Husain-Syed, F. Management of Acute Kidney Injury in Patients with COVID-19. Lancet Respir. Med. 2020, 8, 738–742. [Google Scholar] [CrossRef]
- Li, Z.; Wang, S.; Huo, X.; Yu, H.; Lu, J.; Zhang, S.; Li, X.; Cao, Q.; Li, C.; Guo, M.; et al. Cystatin C Expression Is Promoted by Vegfa Blocking, with Inhibitory Effects on Endothelial Cell Angiogenic Functions Including Proliferation, Migration, and Chorioallantoic Membrane Angiogenesis. J. Am. Heart Assoc. 2018, 7, e009167. [Google Scholar] [CrossRef] [Green Version]
- Zamanian-Azodi, M.; Arjmand, B.; Razzaghi, M.; Tavirani, M.R.; Ahmadzadeh, A.; Rostaminejad, M. Platelet and Haemostasis Are the Main Targets in Severe Cases of COVID-19 Infection; a System Biology Study. Arch. Acad. Emerg. Med. 2021, 9, e27. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV-2 Infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Macpherson, M.E.; Halvorsen, B.; Yndestad, A.; Ueland, T.; Mollnes, T.E.; Berge, R.K.; Rashidi, A.; Otterdal, K.; Gregersen, I.; Kong, X.Y.; et al. Impaired HDL Function Amplifies Systemic Inflammation in Common Variable Immunodeficiency. Sci. Rep. 2019, 9, 9427. [Google Scholar] [CrossRef] [PubMed]
- Allard-Ratick, M.P.; Kindya, B.R.; Khambhati, J.; Engels, M.C.; Sandesara, P.B.; Rosenson, R.S.; Sperling, L.S. HDL: Fact, Fiction, or Function? HDL Cholesterol and Cardiovascular Risk. Eur. J. Prev. Cardiol. 2021, 28, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.A.; Govender, M.A.; Brandenburg, J.T.; Winkler, C.A. Kidney Disease and APOL1. Hum. Mol. Genet. 2021, 30, R129–R137. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Hospitalized | Hospitalized | ICU | EXITUS | ||||||||||||||||
| A. Discovery cohort | <60 | 60–80 | <60 | 60–80 | >80 | <60 | 60–80 | <60 | 60–80 | >80 | |||||||||
| (n = 8) | (n = 8) | (n = 9) | (n = 10) | (n = 9) | (n = 6) | (n = 4) | (n = 2) | (n = 6) | (n = 10) | ||||||||||
| Age (years), mean (SD) | 46 ± 7 | 68 ± 5 | 37 ± 4 | 63 ± 2 | 83 ± 2 | 49 ± 6 | 66 ± 4 | 50 ± 8 | 69 ± 4 | 85 ± 3 | |||||||||
| Male, No. (%) | 3 (38%) | 4 (50%) | 5 (56%) | 7 (70%) | 3 (33%) | 5 (83%) | 3 (75%) | 2 (100%) | 3 (50%) | 3 (30%) | |||||||||
| Days between symptoms onset and plasma extraction, mean (SD) | 7 ± 2 | 9 ± 2 | 9 ± 2 | 9 ± 2 | 7 ± 1 | 9 ± 2 | 8 ± 1 | 9 | 8 ± 2 | 9 ± 3 | |||||||||
| Comorbidity, No. (%) | 5 (63%) | 6 (75%) | 2 (22%) | 7 (70%) | 9 (100%) | 5 (83%) | 3 (75%) | 2 (100%) | 6 (100%) | 9 (90%) | |||||||||
| Pharmacotherapy, No. (%) | 6 (75%) | 6 (75%) | 4 (44%) | 8 (80%) | 9 (100%) | 6 (100%) | 4 (100%) | 0 | 5 (83%) | 8 (80%) | |||||||||
| Non-Hospitalized | Hospitalized | ICU | EXITUS | Discharged (100 days) | |||||||||||||||
| B. Validation cohort | <60 | 60–80 | >80 | <60 | 60–80 | >80 | <60 | 60–80 | <60 | 60–80 | >80 | <60 | 60–80 | >80 | |||||
| (n = 6) | (n = 8) | (n = 3) | (n = 6) | (n = 5) | (n = 5) | (n = 5) | (n = 10) | (n = 4) | (n = 5) | (n = 6) | (n = 7) | (n = 7) | (n = 7) | ||||||
| Age (years), mean (SD) | 44 ± 9 | 68 ± 5 | 88 ± 7 | 43± 9 | 68 ± 6 | 86 ± 3 | 39 ± 7 | 72 ± 5 | 49 ± 9 | 70 ± 6 | 90 ± 4 | 53 ± 5 | 72 ± 6 | 85 ± 4 | |||||
| Male, No. (%) | 1 | 4 | 2 | 3 | 3 | 2 | 4 | 9 | 3 | 1 | 1 | 7 | 4 | 2 | |||||
| (17%) | (50%) | (67%) | (50%) | (60%) | (40%) | (80%) | (90%) | (75%) | (20%) | (17%) | (100%) | (57%) | (29%) | ||||||
| Days between symptoms onset and plasma extraction, mean (SD) | 6 ± 4 | 8 ± 3 | 5 ± 2 | 7 ± 4 | 6 ± 4 | 6 ± 2 | 6 ± 3 | 8 ± 2 | 9 ± 2 | 6 ± 4 | 5 ± 3 | 170 ± 52 | 150 ± 55 | 166 ± 52 | |||||
| Comorbidity, No. (%) | 6 | 7 | 3 | 4 | 4 | 5 | 5 | 9 | 3 | 5 | 6 | 5 | 7 | 7 | |||||
| (100%) | (87%) | (100%) | (67%) | 80%) | (100%) | (100%) | (90%) | (75%) | (100%) | (100%) | (71%) | (100%) | (100%) | ||||||
| Pharmacotherapy, No. (%) | 5 | 4 | 3 | 3 | 4 | 4 | 5 | 8 | 2 | 4 | 4 | 5 | 7 | 7 | |||||
| (83%) | (50%) | (100%) | (50%) | (80%) | (80%) | (100%) | -0,8 | (50%) | (80%) | (67%) | (71%) | (100%) | (100%) | ||||||
| Age (years), mean (SD) | 44 ± 9 | 68 ± 5 | 88 ± 7 | 43± 9 | 68 ± 6 | 86 ± 3 | 39 ± 7 | 72 ± 5 | 49 ± 9 | 70 ± 6 | 90 ± 4 | 53 ± 5 | 72 ± 6 | 85 ± 4 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuñez, E.; Orera, I.; Carmona-Rodríguez, L.; Paño, J.R.; Vázquez, J.; Corrales, F.J. Mapping the Serum Proteome of COVID-19 Patients; Guidance for Severity Assessment. Biomedicines 2022, 10, 1690. https://doi.org/10.3390/biomedicines10071690
Nuñez E, Orera I, Carmona-Rodríguez L, Paño JR, Vázquez J, Corrales FJ. Mapping the Serum Proteome of COVID-19 Patients; Guidance for Severity Assessment. Biomedicines. 2022; 10(7):1690. https://doi.org/10.3390/biomedicines10071690
Chicago/Turabian StyleNuñez, Estefanía, Irene Orera, Lorena Carmona-Rodríguez, José Ramón Paño, Jesús Vázquez, and Fernando J. Corrales. 2022. "Mapping the Serum Proteome of COVID-19 Patients; Guidance for Severity Assessment" Biomedicines 10, no. 7: 1690. https://doi.org/10.3390/biomedicines10071690
APA StyleNuñez, E., Orera, I., Carmona-Rodríguez, L., Paño, J. R., Vázquez, J., & Corrales, F. J. (2022). Mapping the Serum Proteome of COVID-19 Patients; Guidance for Severity Assessment. Biomedicines, 10(7), 1690. https://doi.org/10.3390/biomedicines10071690