
How Chemotherapy Affects the Tumor Immune Microenvironment: A Narrative Review

 ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
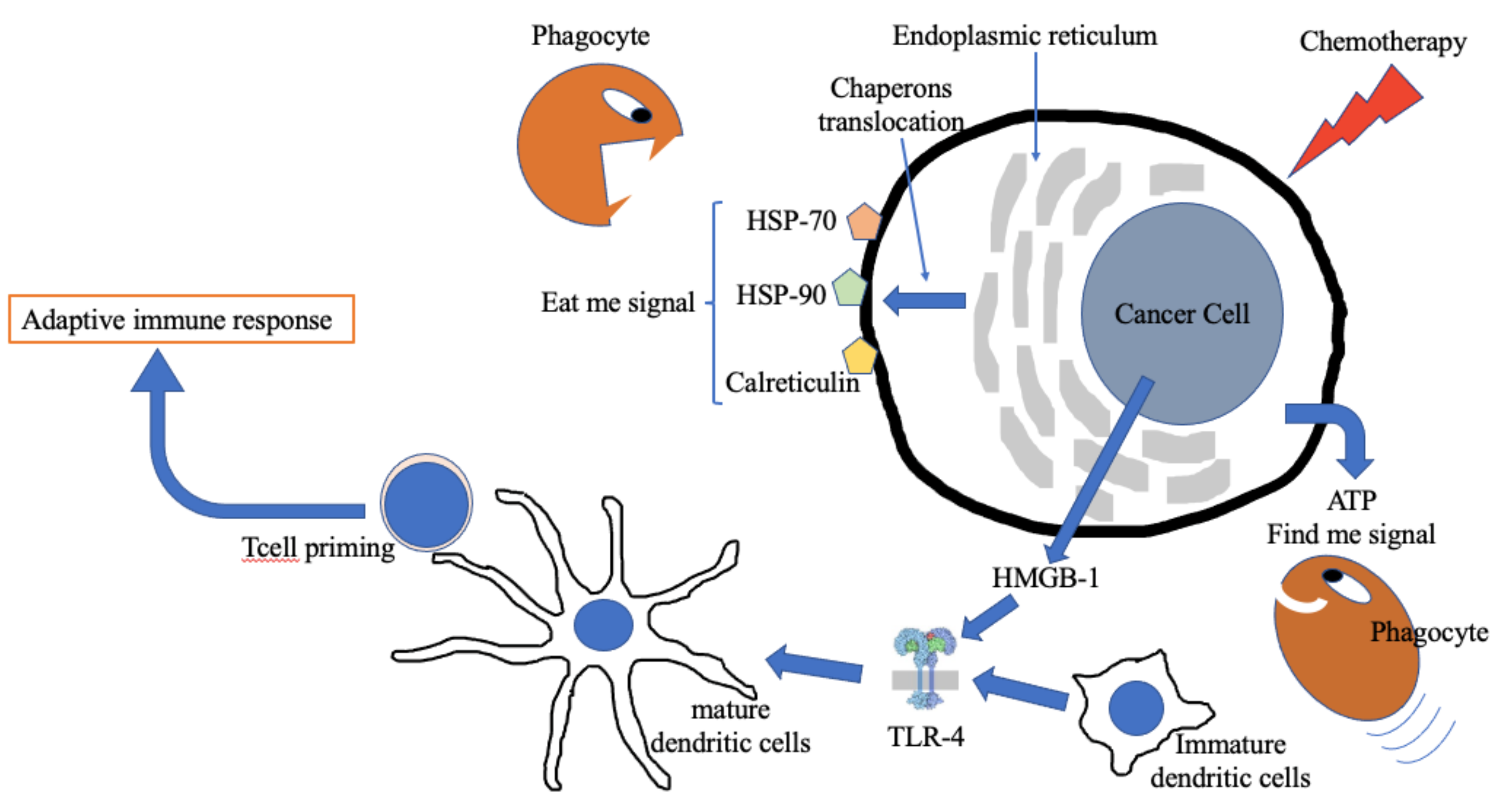
:1. Introduction
2. Chemotherapy
2.1. Targeting Cancer Cells
2.2. Targeting Immune Cells
2.2.1. Inhibition of Immune-Suppressive Cells
- Tregs are among the principal actors in the tumor immune suppressive microenvironment. Indeed, one of the effects associated with the recovery of the antitumor response is the increase in the ratio of effector T cells to regulatory T cells as observed both in humans and in experimental models [12,26].
- MDSCs, a heterogeneous family of immature immune cells, include two main subsets: granulocytic (gMDSC) and monocytic (mMDSC) cells. The former are predominant in humans (about 75% of all MDSCs), while mMDSCs are less represented and more immunosuppressive [29].
- Targeting TAM-M2, the pro-tumor macrophage phenotype, is an important method of chemotherapy to restore the immune response. Chemotherapy may change TAM polarization toward the M1 (the antitumor phenotype), exploiting TAM plasticity.
2.2.2. Activation of Immune Effector Cells
2.3. Effect of Chemotherapy on the Expression of PD-L1
2.4. Targeting the Host Physiology
3. Targeted Therapeutic Drugs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M. State-of-the-art therapeutics: Hodgkin’s lymphoma. J. Clin. Oncol. 2005, 23, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.M. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 2001, 410, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Carbognin, L.; Pilotto, S.; Nortilli, R.; Brunelli, M.; Nottegar, A.; Sperduti, I.; Giannarelli, D.; Bria, E.; Tortora, G. Predictive and Prognostic Role of Tumor-Infiltrating Lymphocytes for Early Breast Cancer According to Disease Subtypes: Sensitivity Analysis of Randomized Trials in Adjuvant and Neoadjuvant Setting. Oncologist 2016, 21, 283–291. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating Immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Devaud, C.; John, L.B.; Westwood, J.A.; Darcy, P. Enhancing immunotherapy using chemotherapy and radiation to modify the tumor microenvironment. Oncoimmunology 2013, 2, e25962. [Google Scholar] [CrossRef]
- Pasquier, E.; Kavallaris, M.; André, N. Metronomic chemotherapy: New rationale for new directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Preissner, K.T.; Fischer, S.; Deindl, E. Extracellular RNA as a Versatile DAMP and Alarm Signal That Influences Leukocyte Recruitment in Inflammation and Infection. Front. Cell Dev. Biol. 2020, 8, 619221. [Google Scholar] [CrossRef]
- Szczesny, B.; Marcatti, M.; Ahmad, A.; Montalbano, M.; Brunyánszki, A.; Bibli, S.-I.; Papapetropoulos, A.; Szabo, C. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci. Rep. 2018, 8, 914. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Messmer, M.N.; Snyder, A.G.; Oberst, A. Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death Differ. 2019, 26, 115–129. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef]
- Lo Nigro, C.; Arnolfo, E.; Taricco, E.; Fruttero, A.; Russi, E.G.; Lucio, F.; Ribero, S.; Comino, A.; Merlano, M.; Ungari, S. The cisplatin–irradiation combination suggests that apoptosis is not a major determinant of clonogenic death. Anti-Cancer Drugs 2007, 18, 659–667. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, M.; Cao, D.; Yang, C.; Jin, J.; Wu, L.; Hong, X.; Li, W.; Lu, L.; Li, J.; et al. ZBP1-MLKL necroptotic signaling potentiates radiation-induced antitumor immunity via intratumoral STING pathway activation. Sci. Adv. 2021, 7, eabf6290. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef]
- Fumet, J.-D.; Limagne, E.; Thibaudin, M.; Ghiringhelli, F. Immunogenic Cell Death and Elimination of Immunosuppressive Cells: A Double-Edged Sword of Chemotherapy. Cancers 2020, 12, 2637. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell. Physiol. 2019, 234, 7983–7993. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Dimeloe, S.; Frick, C.; Fischer, M.; Gubser, P.M.; Razik, L.; Bantug, G.R.; Ravon, M.; Langenkamp, A.; Hess, C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur. J. Immunol. 2014, 44, 3614–3620. [Google Scholar] [CrossRef] [PubMed]
- Mandruzzato, S.; Solito, S.; Falisi, E.; Francescato, S.; Chiarion-Sileni, V.; Mocellin, S.; Zanon, A.; Rossi, C.R.; Nitti, D.; Bronte, V.; et al. IL4Rα+Myeloid-Derived Suppressor Cell Expansion in Cancer Patients. J. Immunol. 2009, 182, 6562–6568. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I–Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef]
- Wesolowski, R.; Duggan, M.C.; Stiff, A.; Markowitz, J.; Trikha, P.; Levine, K.M.; Schoenfield, L.; Abdel-Rasoul, M.; Layman, R.; Ramaswamy, B.; et al. Circulating myeloid-derived suppressor cells increase in patients undergoing neo-adjuvant chemotherapy for breast cancer. Cancer Immunol. Immunother. 2017, 66, 1437–1447. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Alban, T.J.; Grabowski, M.M.; Alvarado, A.G.; Otvos, B.; Bayik, D.; Roversi, G.; McGraw, M.; Huang, P.; Mohammadi, A.M.; et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight 2019, 4, e130748. [Google Scholar] [CrossRef]
- Eriksson, E.; Wenthe, J.; Irenaeus, S.; Loskog, A.; Ullenhag, G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282. [Google Scholar] [CrossRef]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. OncoImmunology 2019, 8, e1596004. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1- profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- Owen, W.; Thurs, K. Basal Cell Carcinoma Presenting as a Nonhealing Wound. Adv. Ski. Wound Care 2009, 22, 353–355. [Google Scholar] [CrossRef]
- Laoui, D.; Van Overmeire, E.; Van Ginderachter, J. Unsuspected allies: Chemotherapy teams up with immunity to fight cancer. Eur. J. Immunol. 2013, 43, 2538–2542. [Google Scholar] [CrossRef]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Kaneno, R.; Shurin, G.V.; Tourkova, I.L.; Shurin, M.R. Chemomodulation of human dendritic cell function by antineoplastic agents in low noncytotoxic concentrations. J. Transl. Med. 2009, 7, 58. [Google Scholar] [CrossRef]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40–CD40L axis in immunity against infection and cancer. ImmunoTargets Ther. 2018, 7, 55–61. [Google Scholar] [CrossRef]
- Pfannenstiel, L.W.; Lam, S.S.; Emens, L.A.; Jaffee, E.M.; Armstrong, T.D. Paclitaxel enhances early dendritic cell maturation and function through TLR4 signaling in mice. Cell. Immunol. 2010, 263, 79–87. [Google Scholar] [CrossRef]
- Yang, R.; Elsaadi, S.; Misund, K.; Abdollahi, P.; Vandsemb, E.N.; Moen, S.H.; Kusnierczyk, A.; Slupphaug, G.; Standal, T.; Waage, A.; et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J. Immunother. Cancer 2019, 8, e000610. [Google Scholar] [CrossRef]
- Augustin, R.C.; Leone, R.D.; Naing, A.; Fong, L.; Bao, R.; Luke, J.J. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J. Immunother. Cancer 2022, 10, e004089. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Liu, X.; Chen, C.; Harrington, S.M.; Cao, S.; Xie, T.; Pham, T.; Mansfield, A.S.; Yan, Y.; et al. Targeting B7-H1 (PD-L1) sensitizes cancer cells to chemotherapy. Heliyon 2018, 4, e01039, Erratum in Heliyon 2019, 5, e01039. [Google Scholar] [CrossRef]
- Chacon, J.A.; Schutsky, K.; Powell, D.J. The Impact of Chemotherapy, Radiation and Epigenetic Modifiers in Cancer Cell Expression of Immune Inhibitory and Stimulatory Molecules and Anti-Tumor Efficacy. Vaccines 2016, 4, 43. [Google Scholar] [CrossRef]
- Park, S.E.; Lee, S.H.; Ahn, J.S.; Ahn, M.-J.; Park, K.; Sun, J.-M. Increased Response Rates to Salvage Chemotherapy Administered after PD-1/PD-L1 Inhibitors in Patients with Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 106–111. [Google Scholar] [CrossRef]
- Denaro, N.; Merlano, M.C. Unexpected response with palliative conventional therapy in head and neck squamous cell carcinoma after anti-programmed death-1 progression. Head Neck 2019, 41, E42–E47. [Google Scholar] [CrossRef]
- Saleh, K.; Daste, A.; Martin, N.; Pons-Tostivint, E.; Auperin, A.; Herrera-Gomez, R.G.; Baste-Rotllan, N.; Bidault, F.; Guigay, J.; Le Tourneau, C.; et al. Response to salvage chemotherapy after progression on immune checkpoint inhibitors in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. Eur. J. Cancer 2019, 121, 123–129. [Google Scholar] [CrossRef]
- Takahashi, T.; Tateishi, A.; Bychkov, A.; Fukuoka, J. Remarkable Alteration of PD-L1 Expression after Immune Checkpoint Therapy in Patients with Non-Small-Cell Lung Cancer: Two Autopsy Case Reports. Int. J. Mol. Sci. 2019, 20, 2578. [Google Scholar] [CrossRef] [PubMed]
- Rojkó, L.; Reiniger, L.; Téglási, V.; Fábián, K.; Pipek, O.; Vágvölgyi, A.; Agócs, L.; Fillinger, J.; Kajdácsi, Z.; Tímár, J.; et al. Chemotherapy treatment is associated with altered PD-L1 expression in lung cancer patients. J. Cancer Res. Clin. Oncol. 2018, 144, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Deleemans, J.M.; Chleilat, F.; Reimer, R.A.; Henning, J.-W.; Baydoun, M.; Piedalue, K.-A.; McLennan, A.; Carlson, L.E. The chemo-gut study: Investigating the long-term effects of chemotherapy on gut microbiota, metabolic, immune, psychological and cognitive parameters in young adult Cancer survivors; study protocol. BMC Cancer 2019, 19, 1243. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Derosa, L.; Hellmann, M.D.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.J.; Arbour, K.C.; Chaft, J.E.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment with Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients with Cancer. JAMA Oncol. 2019, 5, 1774–1778. [Google Scholar] [CrossRef]
- Lee, K.A.; Luong, M.K.; Shaw, H.; Nathan, P.; Bataille, V.; Spector, T.D. The gut microbiome: What the oncologist ought to know. Br. J. Cancer 2021, 125, 1197–1209. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Spencer, C.N.; Gopalakrishnan, V.; McQuade, J.; Andrews, M.C.; Helmink, B.; Khan, M.W.; Sirmans, E.; Haydu, L.; Cogdill, A.; Burton, E.; et al. Abstract 2838: The gut microbiome (GM) and immunotherapy response are influenced by host lifestyle factors. Cancer Res. 2019, 79, 2838–2838. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]
- Lee, K.A.; Shaw, H.M.; Bataille, V.; Nathan, P.; Spector, T.D. Role of the gut microbiome for cancer patients receiving immunotherapy: Dietary and treatment implications. Eur. J. Cancer 2020, 138, 149–155. [Google Scholar] [CrossRef]
- Klein, M.E.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 inhibitors: The mechanism of action may not be as simple as once thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef]
- Petroni, G.; Buque, A.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Immunomodulation by target anticancer agents. Cancer Cells 2021, 39, 310–345. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 inhibitor Abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef]
- Lelliott, E.J.; Sheppard, K.E.; McArthur, G.A. Harnessing the immunotherapeutic potential of CDK4/6 inhibitors in melanoma: Is timing everything? NPJ Precis. Oncol. 2022, 6, 26. [Google Scholar] [CrossRef]
- Reddy, S.; Reuben, A.; Wargo, J.A. Influences of BRAF Inhibitors on the Immune Microenvironment and the Rationale for Combined Molecular and Immune Targeted Therapy. Curr. Oncol. Rep. 2016, 18, 42. [Google Scholar] [CrossRef]
- Wang, D.; Cong, J.; Fu, B.; Zheng, X.; Sun, R.; Tian, Z.; Wei, H. Immunogenic chemotherapy effectively inhibits KRAS-Driven lung cancer. Cancer Lett. 2020, 492, 31–43. [Google Scholar] [CrossRef]
- Ebert, P.J.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3K inhibition iss synergistic and immunogenic in triple negative breast cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef]
- Ferris, R.L.; Lenz, H.-J.; Trotta, A.M.; García-Foncillas, J.; Schulten, J.; Audhuy, F.; Merlano, M.; Milano, G. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: Harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat. Rev. 2018, 63, 48–60. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, J. Immunological effect of tyrosine kinase inhibitors on the tumor immune environment in non-small cell lung cancer (Review). Oncol. Lett. 2022, 23, 165. [Google Scholar] [CrossRef]
- Manzoni, M.; Rovati, B.; Ronzoni, M.; Loupakis, F.; Mariucci, S.; Ricci, V.; Gattoni, E.; Salvatore, L.; Tinelli, C.; Villa, E.; et al. Immunological Effects of Bevacizumab-Based Treatment in Metastatic Colorectal Cancer. Oncology 2010, 79, 187–196. [Google Scholar] [CrossRef]
- Duan, X.L.; Guo, J.P.; Li, F.; Xiu, C.; Wang, H. Sunitinib inhibits PD-L1 expression in osteosarcoma by targeting STAT3 and remodels the immune system in tumor-bearing mice. Futur. Oncol. 2020, 16, 1815–1824. [Google Scholar] [CrossRef]
- Ocadlikova, D.; Lecciso, M.; Broto, J.M.; Scotlandi, K.; Cavo, M.; Curti, A.; Palmerini, E. Sunitinib Exerts In Vitro Immunomodulatory Activity on Sarcomas via Dendritic Cells and Synergizes With PD-1 Blockade. Front. Immunol. 2021, 12, 577766. [Google Scholar] [CrossRef]


{kind=link}
| Drug | CRT Exposure | ATP Release | HMGB1 Release |
|---|---|---|---|
| Cisplatin | - | + | + |
| Carboplatin | ± | + | ± |
| Oxaliplatin | + | + | ? |
| Docetaxel | + | - | + |
| Paclitaxel | + | + | + |
| Immune Cells | Drugs | Effect | Issues | Model | Ref. |
|---|---|---|---|---|---|
| T-reg | mCTX | Depletion | Dose and scheduling dependent | Human | [27] [28] |
| MDSC | Cape Gem | Depletion | MDSC includes gMDSC and mMDSC Selectivity of active drugs is unclear | Human | [32] [33] |
| TAM-M2 | CTX CDDP CBDCA PCTXL | Reprogramming toward M1 phenotype | Human | [34] [35] | |
| Trabectidin | Depletion | [39] | |||
| DC | PCTXL | Maturation | Experimental data | In vivo and in vitro | [42] |
| CD 8+ T cells | Gem | Proliferation | - | Human | [33] |
| Dietary | |
|---|---|
| Diversifying the diet | Consumption of a great variety of different foods |
| Having a high fiber intake | At least 30 g/day |
| Consuming many different plant species | 30 different species/week recommended |
| Advising patients | Against consumption of self-prescribed commercially available probiotic supplements |
| General | |
| Broad-spectrum antibiotics | Treatment, especially one month before starting immunotherapy, should be avoided unless strictly necessary |
| If antibiotics needed | A microbiology consultation should be required to avoid broad-spectrum antibiotics |
| If broad-spectrum antibiotics given within one month from the planned immune treatment | Consider temporarily delaying the start of immunotherapy |
| Target | Agent | Effect | Ref. |
|---|---|---|---|
| CDK 4/6 | Abemaciclib | Improved antigen presentation; pro-inflammatory cytokines release; depleted Treg | [65] |
| Palbociclib | Improved antigen presentation; pro-inflammatory cytokines release; PD-L1 up-regulation; activation of Teff cells; depleted Treg | [66] | |
| Ribociclib | Improved antigen presentation | [66] | |
| BRAF | Dabrafenib | Improved antigen presentation; enhanced Teff functions | [67] |
| Vemurafenib | Improved antigen presentation; enhanced Teff functions | ||
| MEK | Trametinib | Improved antigen presentation; ICD; | [68] |
| Cobimetinib | Activation of Teff cells | [69] | |
| PI3K | Alpelisib | Improved antigen presentation; PD-L1 down-regulation | [70] |
| EGFR | Cetuximab | Improved antigen presentation; ADCC; ICD; TAM-M2 polarization | [71] |
| Gefitinib | Improved antigen presentation; PD-L1 down-regulation; DC activation | [72] | |
| Erlotinib | Improved antigen presentation | ||
| Afatinib | Improved antigen presentation | ||
| HER-2 | Trastuzumab | Improved antigen presentation; DC activation; ADCC; TAM-M2 polarization | [64] |
| Pertuzumab | Improved antigen presentation; ADCC | ||
| VEGF | Bevacizumab | Teff expansion; DC activation | [73] |
| Apatinib | PD-L1 down-regulation | [74] | |
| Sunitinib | Treg depletion; MDSC depletion | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merlano, M.C.; Denaro, N.; Galizia, D.; Ruatta, F.; Occelli, M.; Minei, S.; Abbona, A.; Paccagnella, M.; Ghidini, M.; Garrone, O. How Chemotherapy Affects the Tumor Immune Microenvironment: A Narrative Review. Biomedicines 2022, 10, 1822. https://doi.org/10.3390/biomedicines10081822
Merlano MC, Denaro N, Galizia D, Ruatta F, Occelli M, Minei S, Abbona A, Paccagnella M, Ghidini M, Garrone O. How Chemotherapy Affects the Tumor Immune Microenvironment: A Narrative Review. Biomedicines. 2022; 10(8):1822. https://doi.org/10.3390/biomedicines10081822
Chicago/Turabian StyleMerlano, Marco Carlo, Nerina Denaro, Danilo Galizia, Fiorella Ruatta, Marcella Occelli, Silvia Minei, Andrea Abbona, Matteo Paccagnella, Michele Ghidini, and Ornella Garrone. 2022. "How Chemotherapy Affects the Tumor Immune Microenvironment: A Narrative Review" Biomedicines 10, no. 8: 1822. https://doi.org/10.3390/biomedicines10081822
APA StyleMerlano, M. C., Denaro, N., Galizia, D., Ruatta, F., Occelli, M., Minei, S., Abbona, A., Paccagnella, M., Ghidini, M., & Garrone, O. (2022). How Chemotherapy Affects the Tumor Immune Microenvironment: A Narrative Review. Biomedicines, 10(8), 1822. https://doi.org/10.3390/biomedicines10081822