
Sacubitril/Valsartan and Ivabradine Attenuate Left Ventricular Remodelling and Dysfunction in Spontaneously Hypertensive Rats: Different Interactions with the Renin–Angiotensin–Aldosterone System

 , , , , and
, , , , and 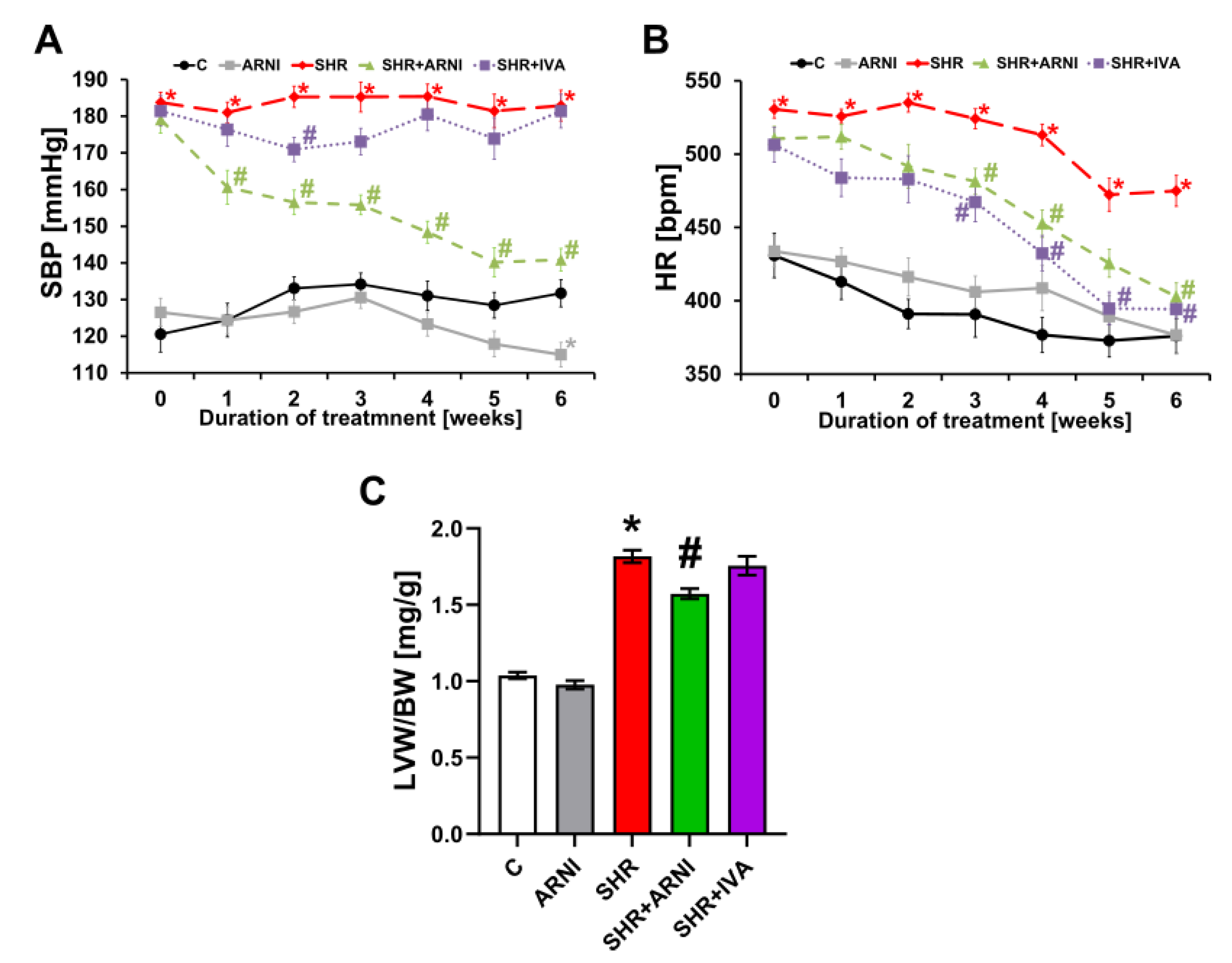{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Determination of Hydroxyproline in the Left Ventricle
2.3. Determination of Serum Angiotensins and Aldosterone Concentration and the Markers of Renin and ACE Activities
2.4. Echocardiography
2.5. Statistical Analysis
3. Results
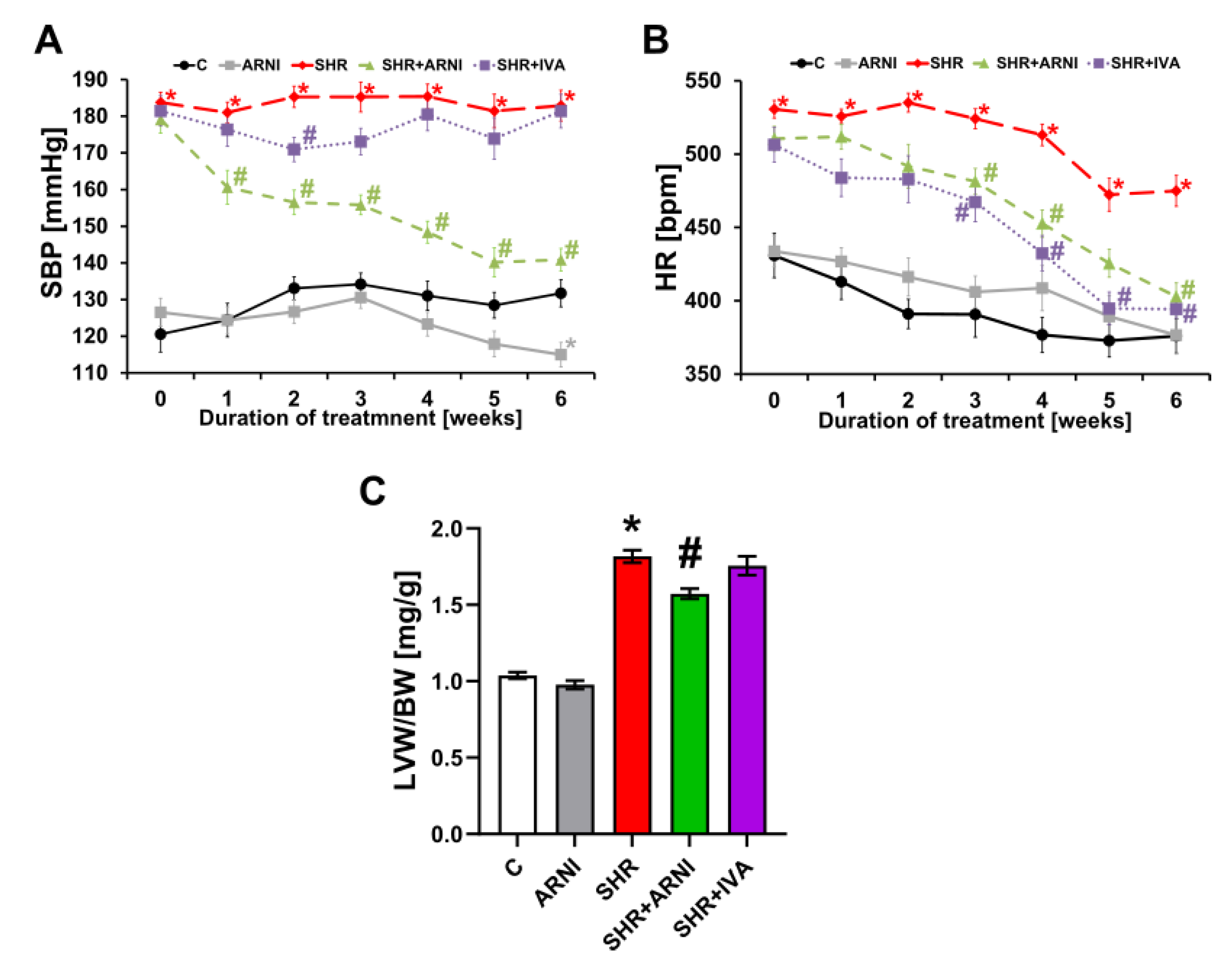
3.1. Haemodynamics and Heart Weights
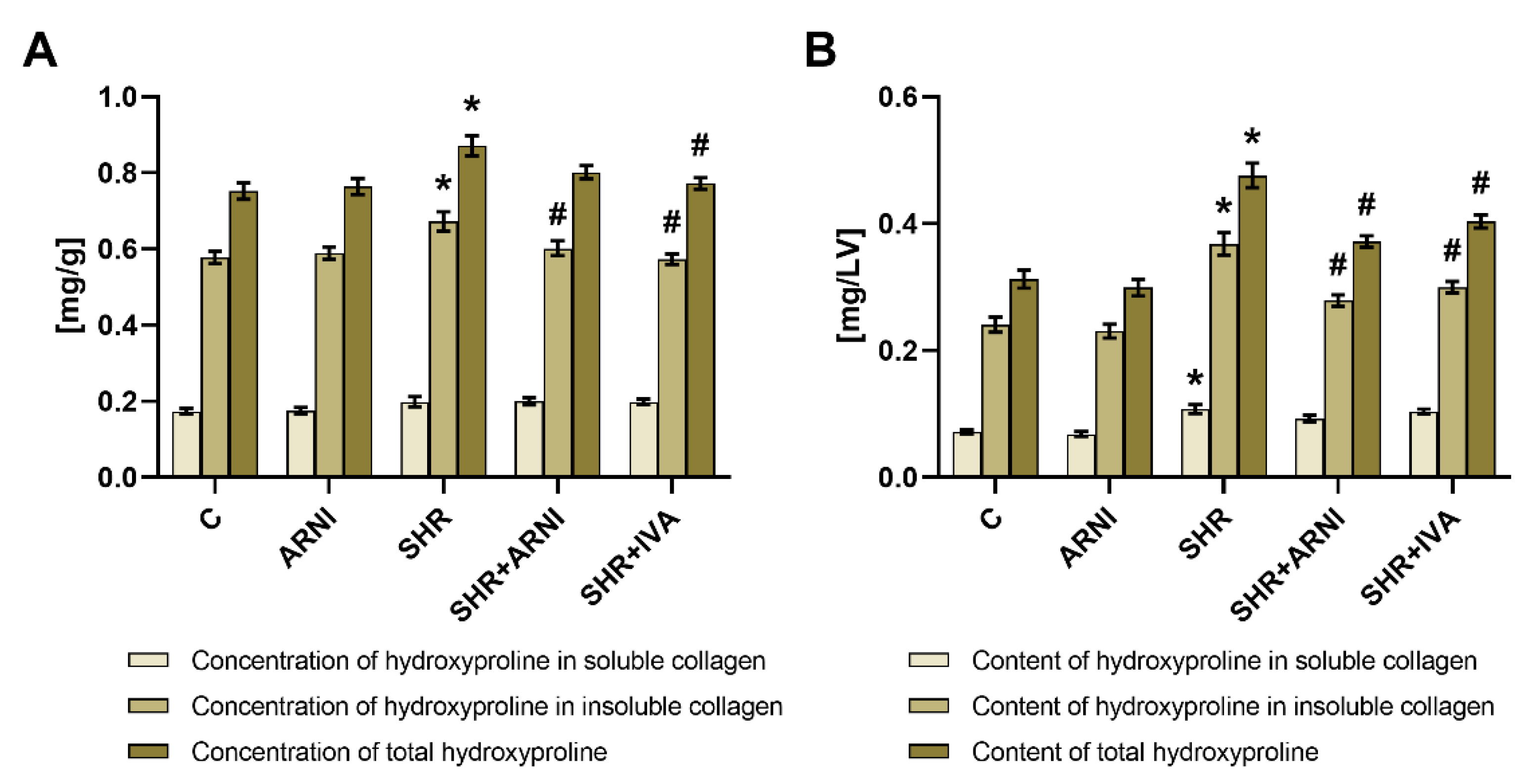
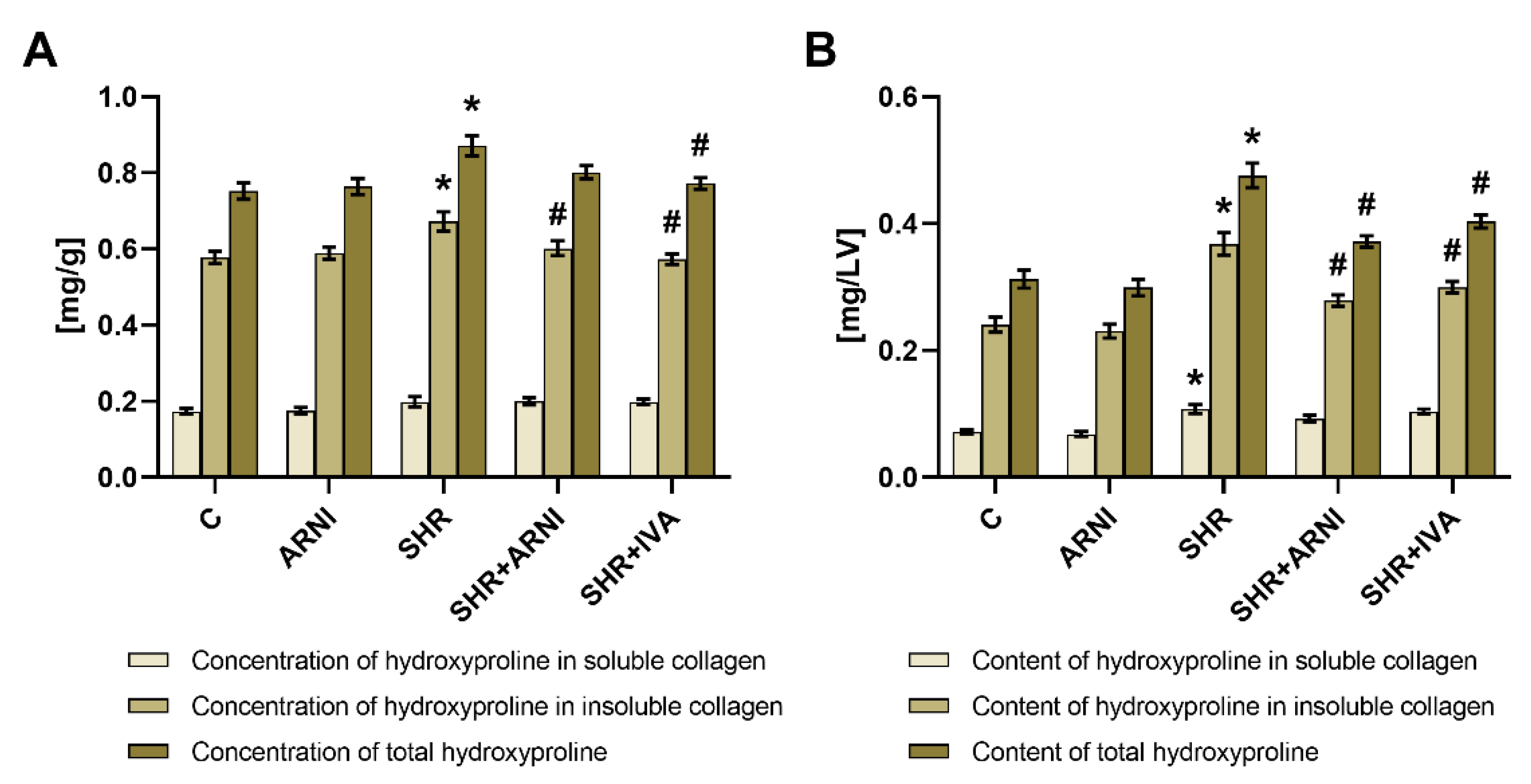
3.2. Hydroxyproline Concentration and Content in Soluble and Insoluble Collagen and Total Hydroxyproline in the Left Ventricle
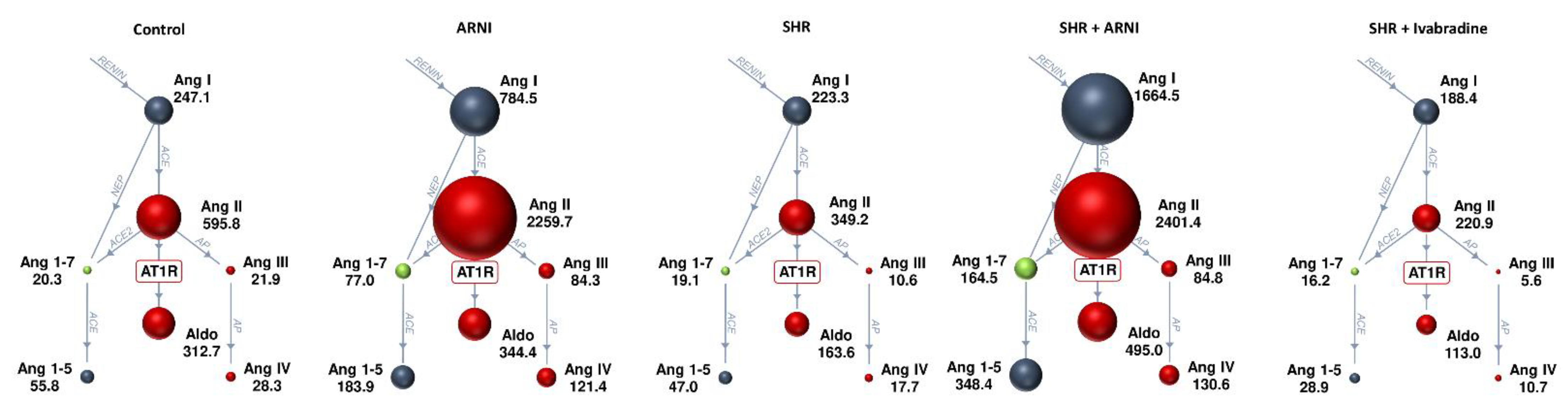
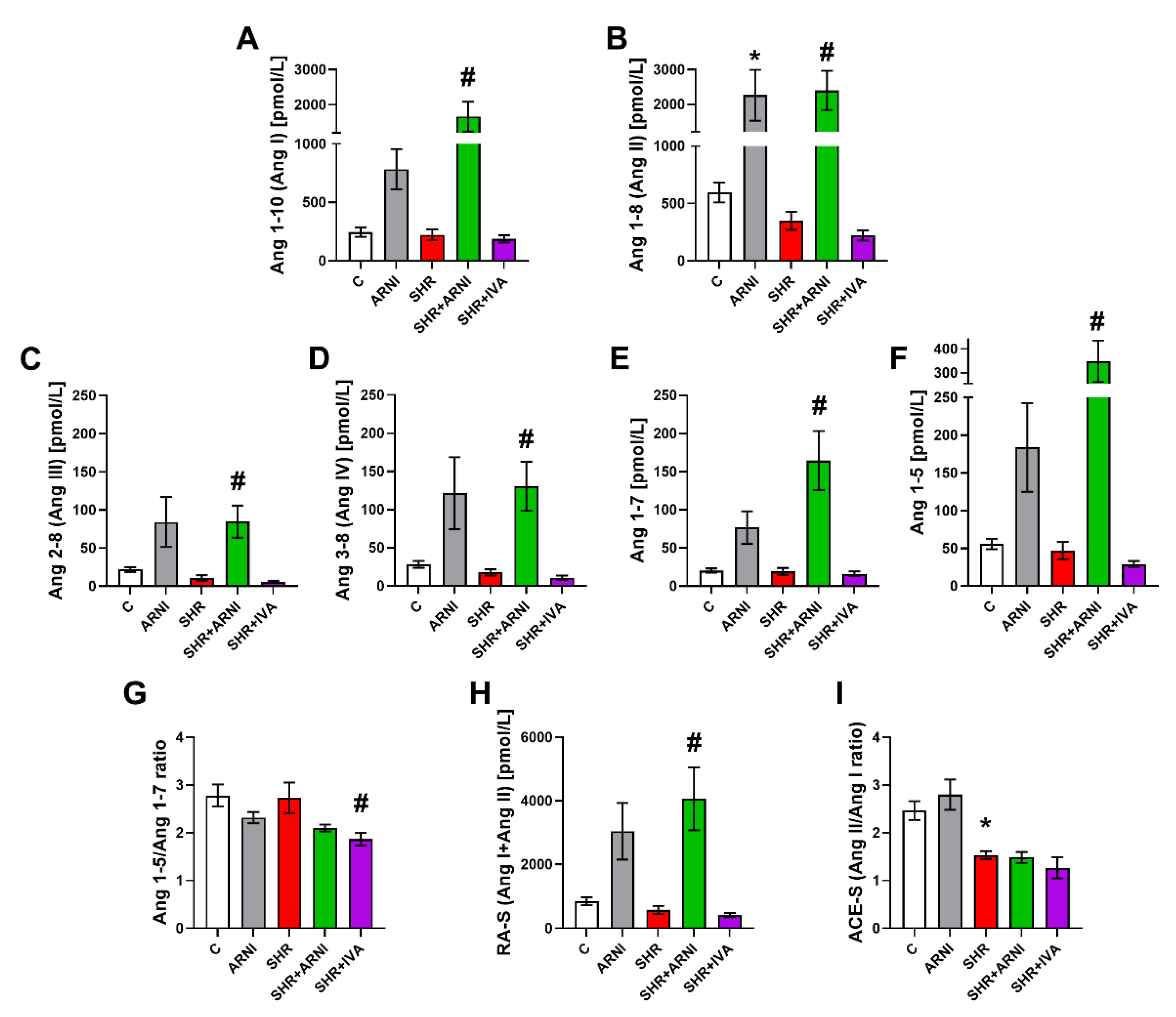
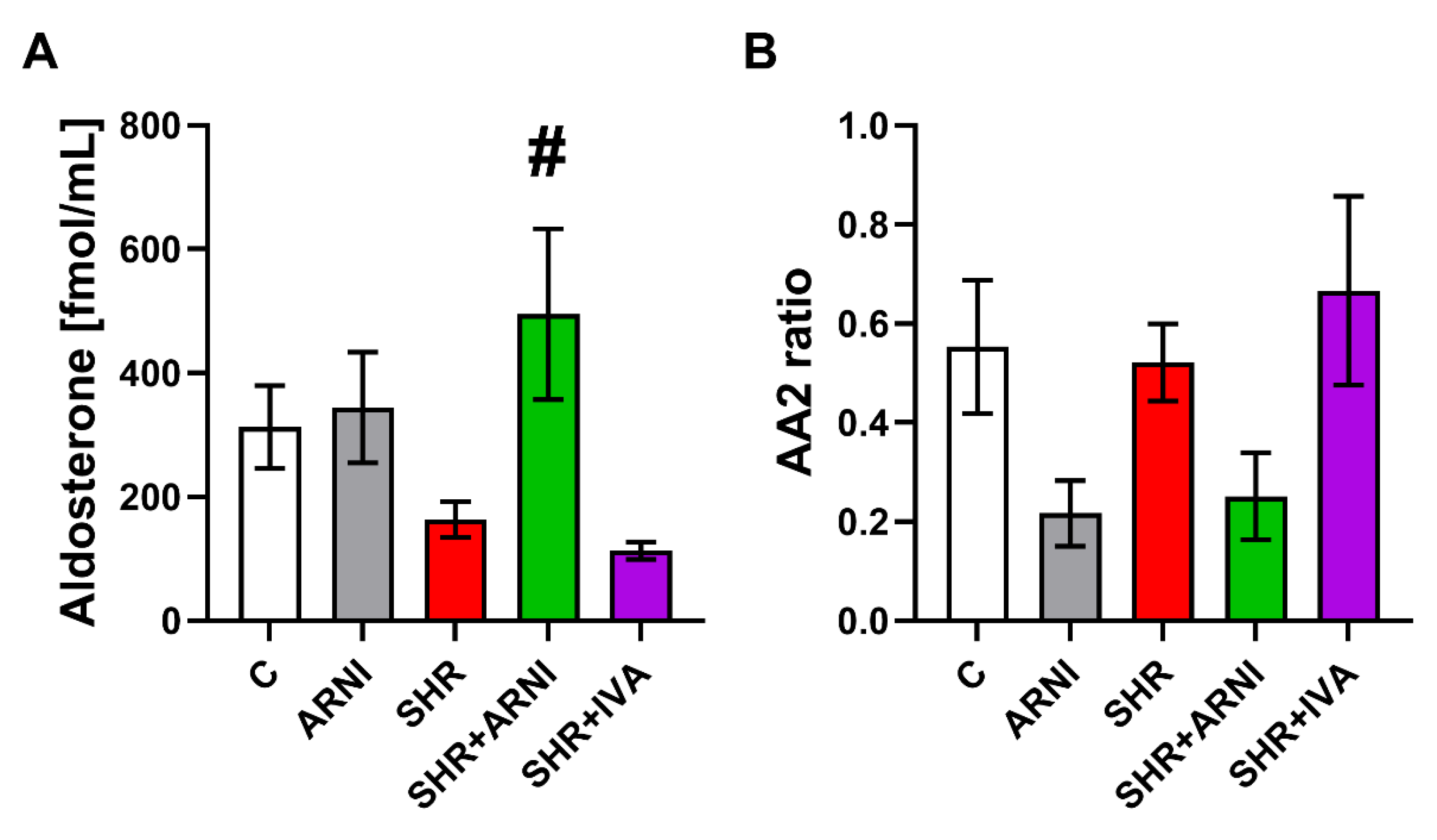
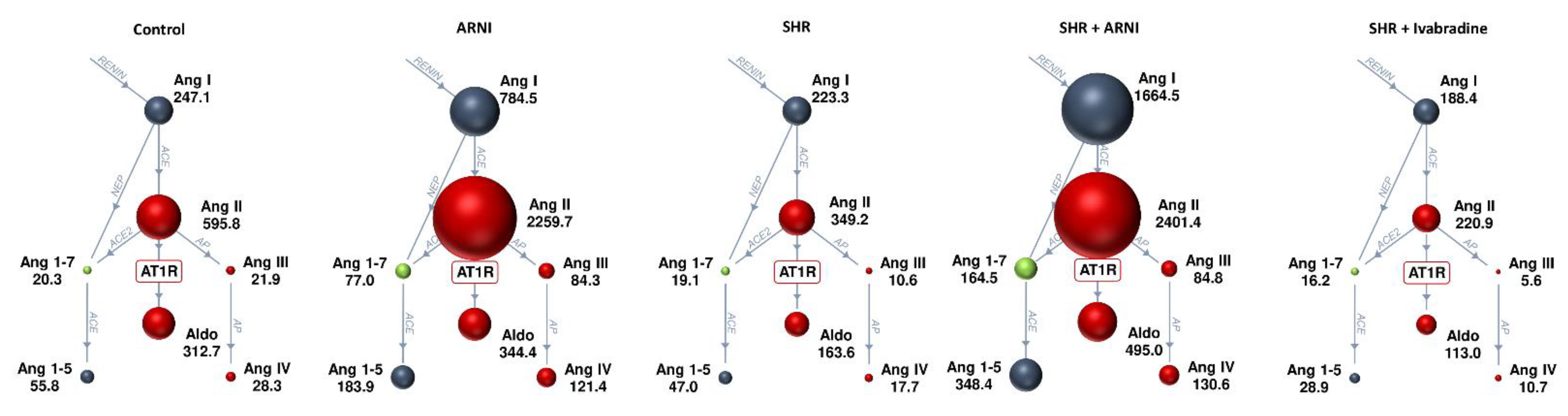
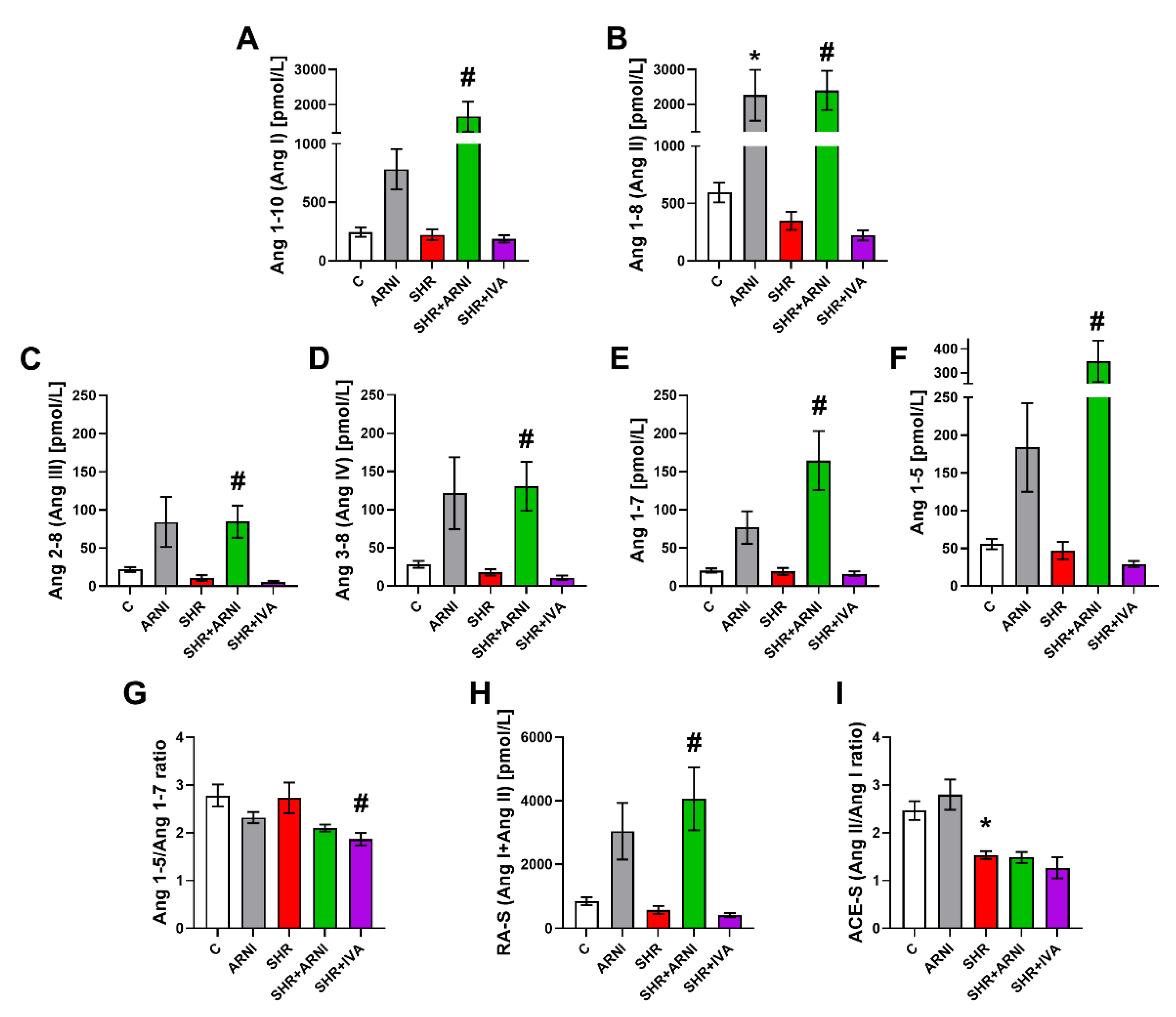
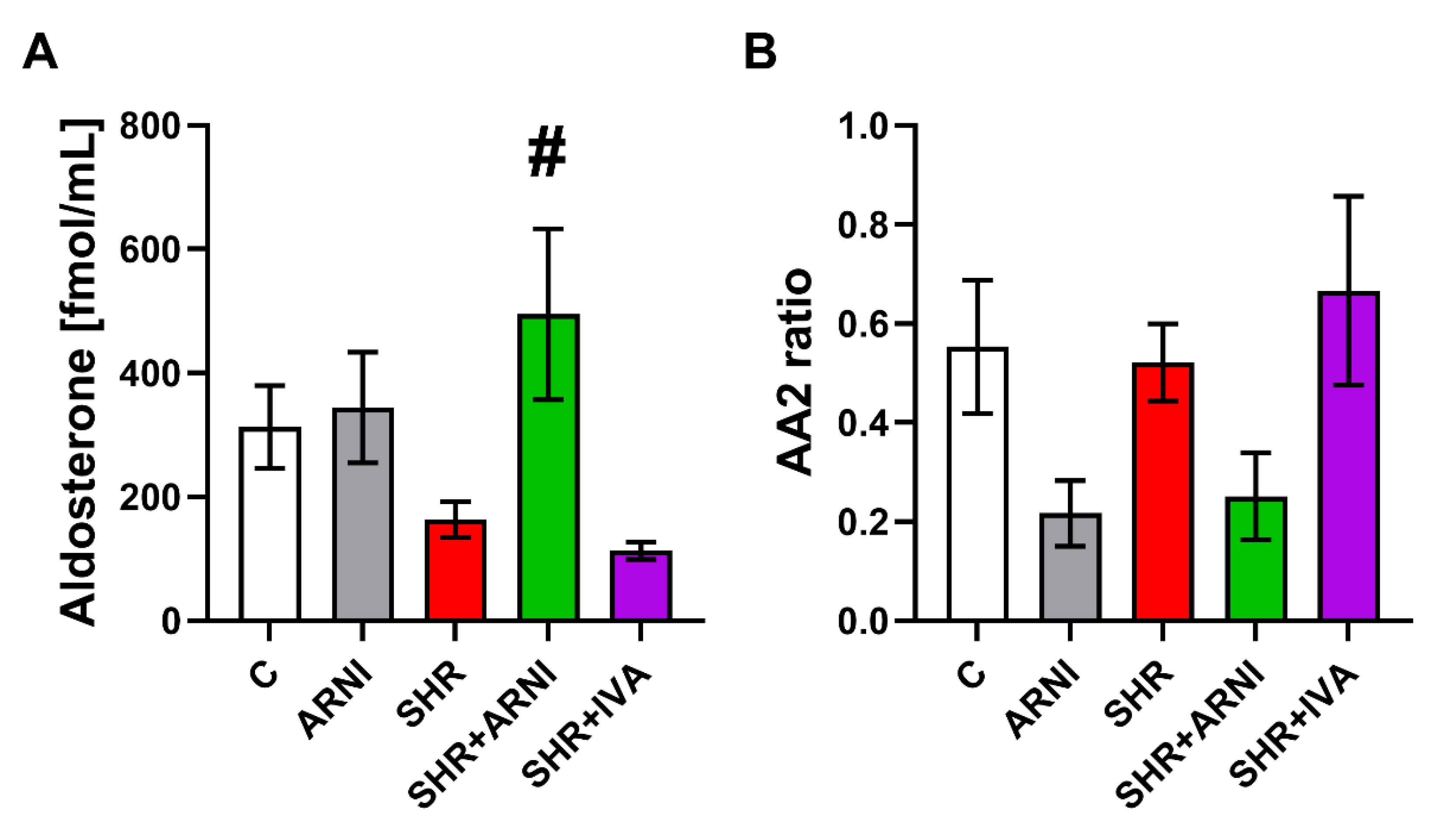
3.3. Serum Concentration of Angiotensins and Aldosterone, and the Markers of Renin and ACE Activities
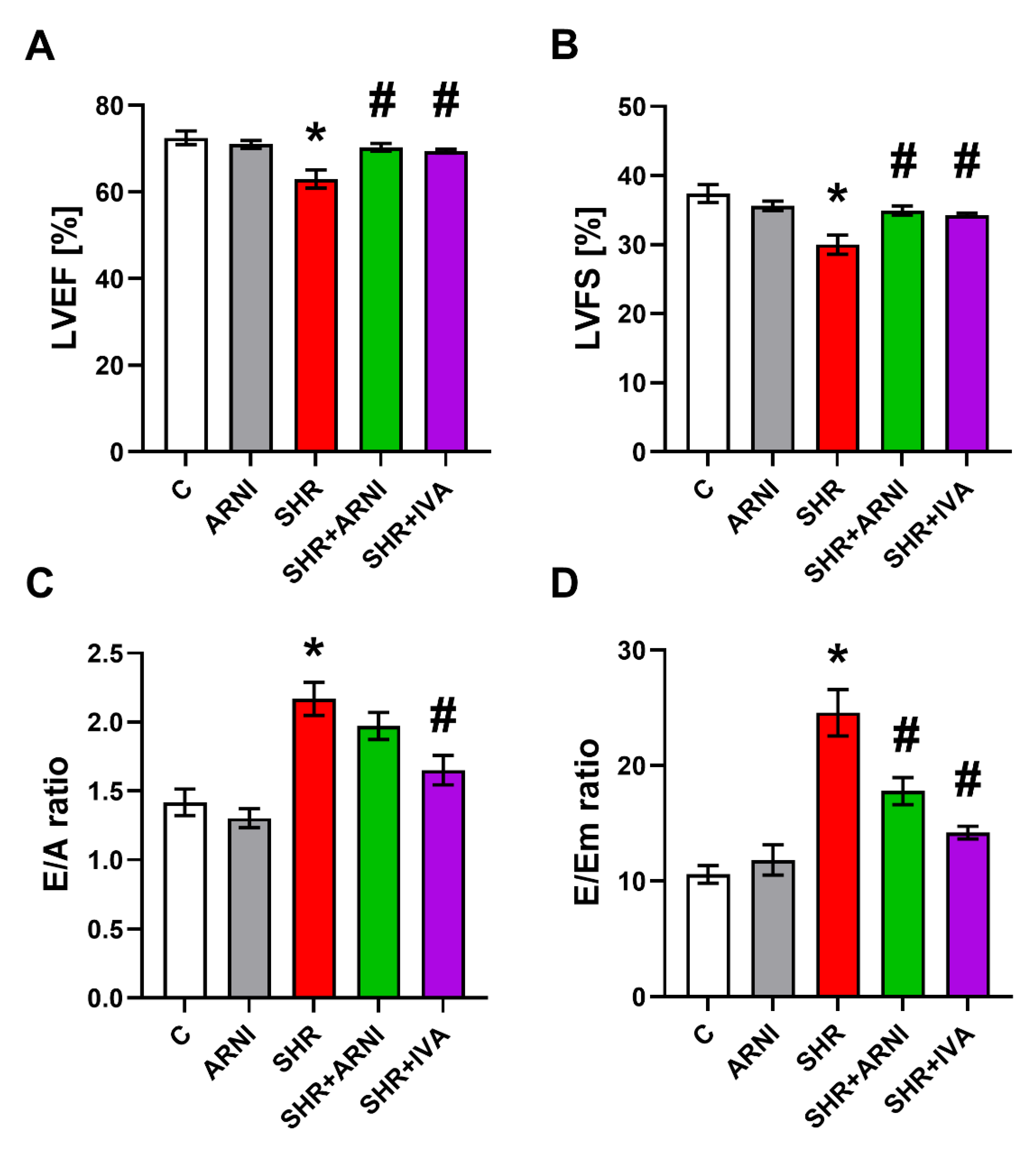
3.4. Echocardiography
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simko, F.; Pechanova, O. Remodelling of the Heart and Vessels in Experimental Hypertension: Advances in Protection. J. Hypertens. 2010, 28 (Suppl. 1), S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. Fibrosis and Hypertensive Heart Disease. Curr. Opin. Cardiol. 2000, 15, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Simko, J. The Potential Role of Nitric Oxide in the Hypertrophic Growth of the Left Ventricle. Physiol. Res. 2000, 49, 37–46. [Google Scholar] [PubMed]
- McMurray, J.J.V. Neprilysin Inhibition to Treat Heart Failure: A Tale of Science, Serendipity, and Second Chances. Eur. J. Heart Fail. 2015, 17, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Dukat, A. Inhibition of Neprilysin—New Horizon in Heart Failure Therapy. Cardiol. Lett. 2016, 25, 273–276. [Google Scholar]
- Packer, M.; McMurray, J.J.V.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin Receptor Neprilysin Inhibition Compared with Enalapril on the Risk of Clinical Progression in Surviving Patients with Heart Failure. Circulation 2015, 131, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieske, B.; Wachter, R.; Shah, S.J.; Baldridge, A.; Szeczoedy, P.; Ibram, G.; Shi, V.; Zhao, Z.; Cowie, M.R.; PARALLAX Investigators and Committee members. Effect of Sacubitril/Valsartan vs Standard Medical Therapies on Plasma NT-ProBNP Concentration and Submaximal Exercise Capacity in Patients with Heart Failure and Preserved Ejection Fraction: The PARALLAX Randomized Clinical Trial. JAMA 2021, 326, 1919–1929. [Google Scholar] [CrossRef]
- Swedberg, K.; Komajda, M.; Böhm, M.; Borer, J.S.; Ford, I.; Dubost-Brama, A.; Lerebours, G.; Tavazzi, L.; SHIFT Investigators. Ivabradine and Outcomes in Chronic Heart Failure (SHIFT): A Randomised Placebo-Controlled Study. Lancet 2010, 376, 875–885. [Google Scholar] [CrossRef]
- Burnett, J.C.; Granger, J.P.; Opgenorth, T.J. Effects of Synthetic Atrial Natriuretic Factor on Renal Function and Renin Release. Am. J. Physiol. 1984, 247, F863–F866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, G.K.; Enwemeka, C.S. A Simplified Method for the Analysis of Hydroxyproline in Biological Tissues. Clin. Biochem. 1996, 29, 225–229. [Google Scholar] [CrossRef]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Goliasch, G.; Wurm, R.; Novak, J.; Strunk, G.; Gyöngyösi, M.; Poglitsch, M.; Säemann, M.D.; Hülsmann, M. Low- and High-Renin Heart Failure Phenotypes with Clinical Implications. Clin. Chem. 2018, 64, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Poglitsch, M.; Cowley, D.; Domenig, O.; McWhinney, B.C.; Ungerer, J.P.J.; Wolley, M.; Stowasser, M. Effects of Ramipril on the Aldosterone/Renin Ratio and the Aldosterone/Angiotensin II Ratio in Patients with Primary Aldosteronism. Hypertension 2020, 76, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Burrello, J.; Buffolo, F.; Domenig, O.; Tetti, M.; Pecori, A.; Monticone, S.; Poglitsch, M.; Mulatero, P. Renin-Angiotensin-Aldosterone System Triple-A Analysis for the Screening of Primary Aldosteronism. Hypertension 2020, 75, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rigel, D.F. Echocardiographic Examination in Rats and Mice. In Cardiovascular Genomics: Methods and Protocols; Methods in Molecular Biology Series; Humana: Totowa, NJ, USA, 2009; Volume 573, pp. 139–155. [Google Scholar] [CrossRef]
- Bernatova, I.; Conde, M.V.; Kopincova, J.; González, M.C.; Puzserova, A.; Arribas, S.M. Endothelial Dysfunction in Spontaneously Hypertensive Rats: Focus on Methodological Aspects. J. Hypertens. Suppl. 2009, 27, S27–S31. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednarova, K.; Adamcova, M.; Paulis, L. Effect of Melatonin, Captopril, Spironolactone and Simvastatin on Blood Pressure and Left Ventricular Remodelling in Spontaneously Hypertensive Rats. J. Hypertens. Suppl. 2009, 27, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Stoyell-Conti, F.F.; Chabbra, A.; Puthentharayil, J.; Rigatto, K.; Speth, R.C. Chronic Administration of Pharmacological Doses of Angiotensin 1-7 and Iodoangiotensin 1-7 Has Minimal Effects on Blood Pressure, Heart Rate, and Cognitive Function of Spontaneously Hypertensive Rats. Physiol. Rep. 2021, 9, e14812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Tang, H.; Sun, S.; Luo, Y.; Ren, X.; Chen, A.; Xu, Y.; Li, P.; Han, Y. Angiotensin-(1-7) Induced Vascular Relaxation in Spontaneously Hypertensive Rats. Nitric Oxide Biol. Chem. 2019, 88, 1–9. [Google Scholar] [CrossRef]
- Jing, Y.; Hu, J.; Zhao, J.; Yang, J.; Huang, N.; Song, P.; Xu, J.; Zhang, M.; Li, P.; Yin, Y. Experimental Study of Blood Pressure and Its Impact on Spontaneous Hypertension in Rats with Xin Mai Jia. Biomed. Pharmacother. 2019, 112, 108689. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Y.; Wang, S.; Wang, Z.; Sun, H.; He, Y.; Yao, W. Effects of Eplerenone on Cerebral Aldosterone Levels and Brain Lesions in Spontaneously Hypertensive Rats. Clin. Exp. Hypertens. 2020, 42, 531–538. [Google Scholar] [CrossRef]
- Tanaka, S.; Ueno, T.; Tsunemi, A.; Nagura, C.; Tahira, K.; Fukuda, N.; Soma, M.; Abe, M. The Adrenal Gland Circadian Clock Exhibits a Distinct Phase Advance in Spontaneously Hypertensive Rats. Hypertens. Res. 2019, 42, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, Z. Historic Perspectives and Recent Advances in Major Animal Models of Hypertension. Acta Pharmacol. Sin. 2005, 26, 295–301. [Google Scholar] [CrossRef]
- Qin, F.; Li, J.; Dai, Y.-F.; Zhong, X.-G.; Pan, Y.-J. Renal Denervation Inhibits the Renin-Angiotensin-Aldosterone System in Spontaneously Hypertensive Rats. Clin. Exp. Hypertens. 2022, 44, 83–92. [Google Scholar] [CrossRef]
- Wang, M.; Han, W.; Zhang, M.; Fang, W.; Zhai, X.; Guan, S.; Qu, X. Long-Term Renal Sympathetic Denervation Ameliorates Renal Fibrosis and Delays the Onset of Hypertension in Spontaneously Hypertensive Rats. Am. J. Transl. Res. 2018, 10, 4042–4053. [Google Scholar] [PubMed]
- Lee, Y.H.; Kim, Y.G.; Moon, J.-Y.; Kim, J.S.; Jeong, K.-H.; Lee, T.W.; Ihm, C.-G.; Lee, S.H. Genetic Variations of Tyrosine Hydroxylase in the Pathogenesis of Hypertension. Electrolyte Blood Press. EBP 2016, 14, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Brocca, M.E.; Pietranera, L.; de Kloet, E.R.; De Nicola, A.F. Mineralocorticoid Receptors, Neuroinflammation and Hypertensive Encephalopathy. Cell. Mol. Neurobiol. 2019, 39, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, X.; Zhang, C.; Zhang, C.; Bu, P. Lcz696 Alleviates Myocardial Fibrosis after Myocardial Infarction through the SFRP-1/Wnt/β-Catenin Signaling Pathway. Front. Pharmacol. 2021, 12, 724147. [Google Scholar] [CrossRef]
- Kompa, A.R.; Lu, J.; Weller, T.J.; Kelly, D.J.; Krum, H.; von Lueder, T.G.; Wang, B.H. Angiotensin Receptor Neprilysin Inhibition Provides Superior Cardioprotection Compared to Angiotensin Converting Enzyme Inhibition after Experimental Myocardial Infarction. Int. J. Cardiol. 2018, 258, 192–198. [Google Scholar] [CrossRef] [PubMed]
- von Lueder, T.G.; Wang, B.H.; Kompa, A.R.; Huang, L.; Webb, R.; Jordaan, P.; Atar, D.; Krum, H. Angiotensin Receptor Neprilysin Inhibitor LCZ696 Attenuates Cardiac Remodeling and Dysfunction after Myocardial Infarction by Reducing Cardiac Fibrosis and Hypertrophy. Circ. Heart Fail. 2015, 8, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suematsu, Y.; Miura, S.-I.; Goto, M.; Matsuo, Y.; Arimura, T.; Kuwano, T.; Imaizumi, S.; Iwata, A.; Yahiro, E.; Saku, K. LCZ696, an Angiotensin Receptor-Neprilysin Inhibitor, Improves Cardiac Function with the Attenuation of Fibrosis in Heart Failure with Reduced Ejection Fraction in Streptozotocin-Induced Diabetic Mice. Eur. J. Heart Fail. 2016, 18, 386–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, K.; Kuwano, T.; Ideishi, A.; Morita, H.; Idemoto, Y.; Goto, M.; Suematsu, Y.; Miura, S.-I. Sacubitril/Valsartan Inhibits Cardiomyocyte Hypertrophy in Angiotensin II-Induced Hypertensive Mice Independent of a Blood Pressure-Lowering Effect. Cardiol. Res. 2020, 11, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, J.; Fu, Y.; Ji, H.; Fang, Z.; Zhou, W.; Fan, H.; Zhang, Y.; Liao, Y.; Yang, T.; et al. Sacubitril/Valsartan Reduces Fibrosis and Alleviates High-Salt Diet-Induced HFpEF in Rats. Front. Pharmacol. 2020, 11, 600953. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Cotugno, M.; Forte, M.; Stanzione, R.; Bianchi, F.; Madonna, M.; Marchitti, S.; Volpe, M. Effects of Dual Angiotensin Type 1 Receptor/Neprilysin Inhibition vs. Angiotensin Type 1 Receptor Inhibition on Target Organ Injury in the Stroke-Prone Spontaneously Hypertensive Rat. J. Hypertens. 2018, 36, 1902–1914. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, R.; Lu, C.; Chen, Q.; Xu, T.; Li, D. Effects of the Angiotensin-Receptor Neprilysin Inhibitor on Cardiac Reverse Remodeling: Meta-Analysis. J. Am. Heart Assoc. 2019, 8, e012272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steckelings, U.M.; Sumners, C. Correcting the Imbalanced Protective RAS in COVID-19 with Angiotensin AT2-Receptor Agonists. Clin. Sci. 2020, 134, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Hrenak, J.; Simko, F. Renin-Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8038. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T. Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers: Potential Allies in the COVID-19 Pandemic Instead of a Threat? Clin. Sci. 2021, 135, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Hrenak, J.; Adamcova, M.; Paulis, L. Renin-Angiotensin-Aldosterone System: Friend or Foe-The Matter of Balance. Insight on History, Therapeutic Implications and COVID-19 Interactions. Int. J. Mol. Sci. 2021, 22, 3217. [Google Scholar] [CrossRef]
- Wang, K.; Basu, R.; Poglitsch, M.; Bakal, J.A.; Oudit, G.Y. Elevated Angiotensin 1-7/Angiotensin II Ratio Predicts Favorable Outcomes in Patients with Heart Failure. Circ. Heart Fail. 2020, 13, e006939. [Google Scholar] [CrossRef] [PubMed]
- Mulrow, P.J. Angiotensin II and Aldosterone Regulation. Regul. Pept. 1999, 80, 27–32. [Google Scholar] [CrossRef]
- Rebuffat, P.; Malendowicz, L.K.; Nussdorfer, G.G.; Mazzocchi, G. Stimulation of Endogenous Nitric Oxide Production Is Involved in the Inhibitory Effect of Adrenomedullin on Aldosterone Secretion in the Rat. Peptides 2001, 22, 923–926. [Google Scholar] [CrossRef]
- Repova, K.; Aziriova, S.; Krajcirovicova, K.; Simko, F. Cardiovascular Therapeutics: A New Potential for Anxiety Treatment? Med. Res. Rev. 2022, 42, 1202–1245. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Baka, T.; Paulis, L.; Reiter, R.J. Elevated Heart Rate and Nondipping Heart Rate as Potential Targets for Melatonin: A Review. J. Pineal Res. 2016, 61, 127–137. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T. Ivabradine and Blood Pressure Reduction: Underlying Pleiotropic Mechanisms and Clinical Implications. Front. Cardiovasc. Med. 2021, 8, 607998. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, Z.; Li, B.; Wang, Z.; Chen, S. Ivabradine Improved Left Ventricular Function and Pressure Overload-Induced Cardiomyocyte Apoptosis in a Transverse Aortic Constriction Mouse Model. Mol. Cell. Biochem. 2019, 450, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Baka, T.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Paulis, L.; Adamcova, M. Ivabradine Improves Survival and Attenuates Cardiac Remodeling in Isoproterenol-Induced Myocardial Injury. Fundam. Clin. Pharmacol. 2021, 35, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Baka, T.; Poglitsch, M.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Zorad, S.; Adamcova, M.; Paulis, L. Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. Int. J. Mol. Sci. 2018, 19, 3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondicova, K.; Hegedusova, N.; Tibensky, M.; Mravec, B. Ivabradine Reduces Baseline and Stress-Induced Increase of Heart Rate and Blood Pressure and Modulates Neuroendocrine Stress Response in Rats Depending on Stressor Intensity. Gen. Physiol. Biophys. 2019, 38, 165–173. [Google Scholar] [CrossRef]
- El-Naggar, A.E.; El-Gowilly, S.M.; Sharabi, F.M. Possible Ameliorative Effect of Ivabradine on the Autonomic and Left Ventricular Dysfunction Induced by Doxorubicin in Male Rats. J. Cardiovasc. Pharmacol. 2018, 72, 22–31. [Google Scholar] [CrossRef]
- Böhm, M.; Borer, J.S.; Camm, J.; Ford, I.; Lloyd, S.M.; Komajda, M.; Tavazzi, L.; Talajic, M.; Lainscak, M.; Reil, J.-C.; et al. Twenty-Four-Hour Heart Rate Lowering with Ivabradine in Chronic Heart Failure: Insights from the SHIFT Holter Substudy. Eur. J. Heart Fail. 2015, 17, 518–526. [Google Scholar] [CrossRef]
- van Thiel, B.S.; Góes Martini, A.; Te Riet, L.; Severs, D.; Uijl, E.; Garrelds, I.M.; Leijten, F.P.J.; van der Pluijm, I.; Essers, J.; Qadri, F.; et al. Brain Renin-Angiotensin System: Does It Exist? Hypertension 2017, 69, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Raizada, V.; Skipper, B.; Luo, W.; Griffith, J. Intracardiac and Intrarenal Renin-Angiotensin Systems: Mechanisms of Cardiovascular and Renal Effects. J. Investig. Med. 2007, 55, 341–359. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simko, F.; Baka, T.; Stanko, P.; Repova, K.; Krajcirovicova, K.; Aziriova, S.; Domenig, O.; Zorad, S.; Adamcova, M.; Paulis, L. Sacubitril/Valsartan and Ivabradine Attenuate Left Ventricular Remodelling and Dysfunction in Spontaneously Hypertensive Rats: Different Interactions with the Renin–Angiotensin–Aldosterone System. Biomedicines 2022, 10, 1844. https://doi.org/10.3390/biomedicines10081844
Simko F, Baka T, Stanko P, Repova K, Krajcirovicova K, Aziriova S, Domenig O, Zorad S, Adamcova M, Paulis L. Sacubitril/Valsartan and Ivabradine Attenuate Left Ventricular Remodelling and Dysfunction in Spontaneously Hypertensive Rats: Different Interactions with the Renin–Angiotensin–Aldosterone System. Biomedicines. 2022; 10(8):1844. https://doi.org/10.3390/biomedicines10081844
Chicago/Turabian StyleSimko, Fedor, Tomas Baka, Peter Stanko, Kristina Repova, Kristina Krajcirovicova, Silvia Aziriova, Oliver Domenig, Stefan Zorad, Michaela Adamcova, and Ludovit Paulis. 2022. "Sacubitril/Valsartan and Ivabradine Attenuate Left Ventricular Remodelling and Dysfunction in Spontaneously Hypertensive Rats: Different Interactions with the Renin–Angiotensin–Aldosterone System" Biomedicines 10, no. 8: 1844. https://doi.org/10.3390/biomedicines10081844