
Internal Ribosome Entry Site (IRES)-Mediated Translation and Its Potential for Novel mRNA-Based Therapy Development


Abstract
:1. Introduction
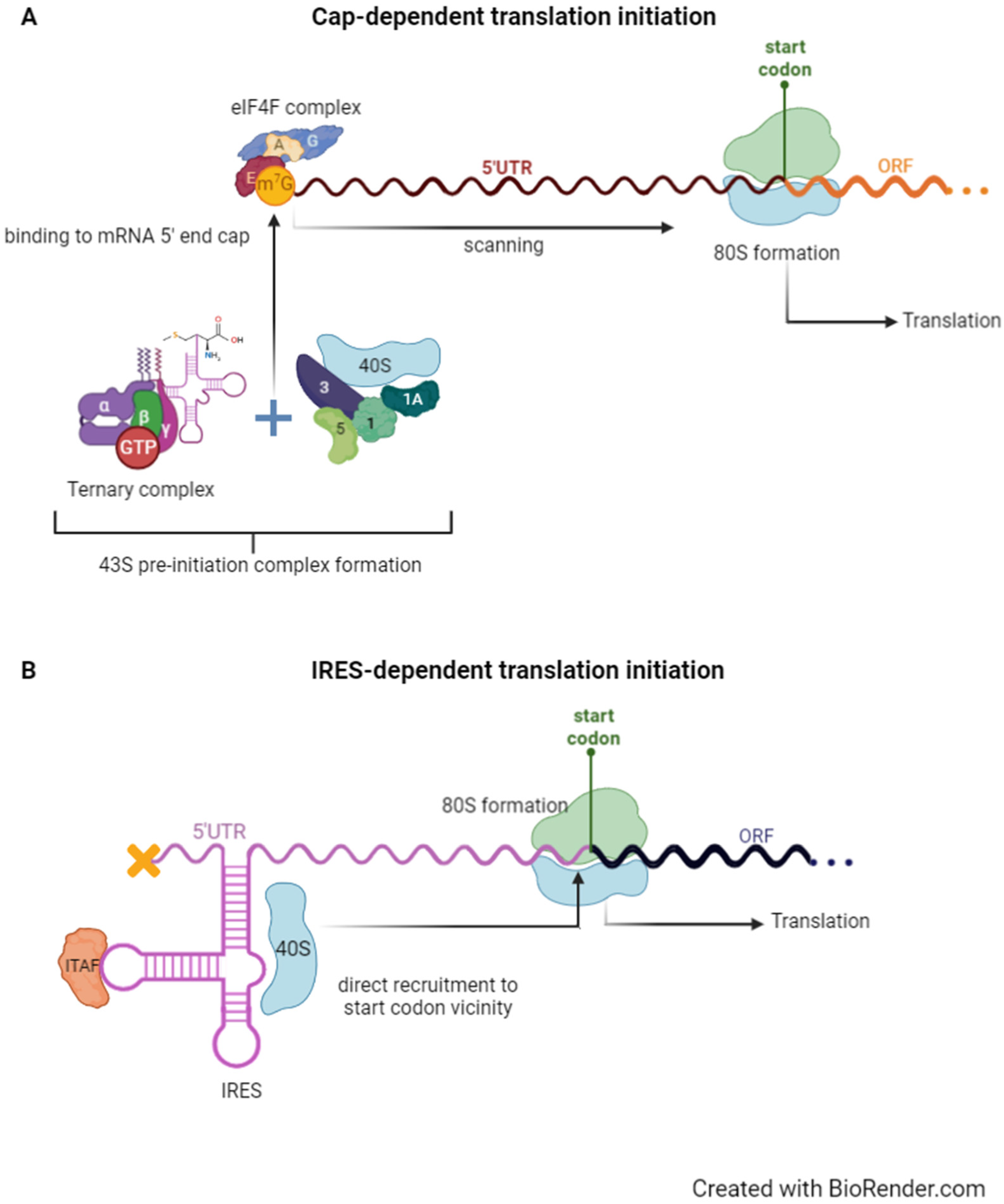
2. Cap- Versus IRES-Dependent Translation Initiation
2.1. Canonical 5′ Cap-Dependent Translation Initiation
2.2. IRES-Dependent Translation Initiation and Its Regulation by IRES Trans-Acting Factors
2.2.1. IRES-Dependent Translation of Circular RNAs (circRNA)
2.2.2. IRES-Mediated Translation of Different Protein Isoforms from Monocistronic Genes
2.2.3. IRES-Mediated Translation of Polycistronic Genes
- (i)
- a single transcript that coordinately expresses at least two protein subunits that are part of a multi-subunit complex. This is the case of tenocyclidine [1-(1-(2-thienyl)cyclohexyl)piperidine binding protein (TCP-BP), which is present in rat brain synaptic membranes and binds glutamate agonists. It is composed of two subunits, PRO-1 and PRO-2; the former is cap-dependently translated, whereas the latter is IRES-dependently translated through an element occurring in the intercistronic region [43,51];
- (ii)
- a single transcript that encodes different protein products with similar structure and function that are differentially expressed, i.e., transcripts that include two cistrons, one encoding a primary protein expressed through a cap-dependent translation mechanism and another encoding a secondary protein translated through a cap-independent mechanism. An example of such a transcript is the free fatty acid receptor 1 (FFAR1), which encodes the G-protein receptor (GPR) 40 and the GPR41 [52]. GPR40 is a receptor for long chain fatty acids, whereas GPR41 is activated by short chain fatty acids [43,53]. This cistronic organisation accounts for coordinated regulation of both receptors. Another example that fits in this group is the meloe mRNA. This is a polycistronic transcript responsible for expressing the melanoma antigens MELOE-1 and MELOE-2, which contain functional IRESs to mediate the expression of such proteins [54]. IRES-mediated translation accounts for the selective expression of these two proteins in melanoma cells, rather than in normal melanocytes [54]. Charpentier et al. identified MELOE-3, a protein with poor immunogenicity encoded by an additional ORF in the 5′ UTR of meloe and translated by the cap-dependent mechanism, reinforcing the importance of targeting MELOE-1 and MELOE-2 IRES-dependent translation for melanoma immunotherapy [43,55];
- (iii)
- a single transcript that encodes functionally distinct proteins whose expression is programmatically related, meaning two proteins that function differentially but play a role in the same pathway [43], like the PITSLRE/CDK11 duplicate genes CdcL1 and CdcL2. Each one encodes two cyclin-dependent protein kinase isoforms, p110 and p58, of which the p58 is IRES-translated [56]. This IRES-dependent translation is cell cycle-dependent and allows translation of p58 during the G2/M transition [43,56]. Also, the voltage-gated Ca2+ channel (CACNA1A) mRNA is bicistronic and encodes both the normal-length α1A subunit (wild-type transcription factor α1 antichymotrypsin, α1ACT) and the expanded polyQ tract subunit (extended α1ACT). The latter is an IRES-translated protein from at least one spliced form of the same CACNA1A mRNA [57]. The myotrophin (MTPN) gene is also transcribed into an mRNA with two adjacent tandem ORFs. These ORFs express two proteins—myotrophin, translated through the cap-dependent mechanism, and autosomal dominant adult-onset distal myopathy-6 (MPD6), translated through an IRES element [58]. Similar to what happens to CACNA1A, the proteins encoded by MTPN have distinct roles, but are programmatically related [43]—myotrophin works in the dimerization of NFκB in cardiac tissue and MPD6 is associated with the immune response in some types of cancer [58];
- (iv)
- a single transcript that encodes proteins produced by stimulus-coupled protease cleavage or by IRES-dependent translation initiation [43]. This is the case of transcripts with two overlapping ORFs that code products required for signal transduction, in which the first cistron codes for a receptor initiating signal transduction upon ligand binding, whereas the downstream cistron produces a constitutively active signal [43]. Notch2, for instance, is a gene encoding a receptor involved in the ligand-receptor notch-signalling pathway [59]. The interaction of Notch2 with the extracellular notch ligand triggers the protease cleavage of the C-terminal polypeptide, the notch intracellular domain (NICD) [60]. Notch2-ICD is translated via an IRES occurring in the Notch2 coding region [60]. Another example is the Her2 gene, a tyrosine kinase receptor involved in cancers and neurodegenerative diseases. It is a polycistronic gene encoding the full-length HER2 protein and several C-terminal fragments (CTF) [61]. These CTFs are translated through IRESs within the HER2 coding region [43,61];
- (v)
- a single transcript with different ORFs separated by IRES-containing intercistronic regions. This is the case of the tricistronic c-myc mRNA that, when transcribed from the alternative upstream promoter P0, contains three different ORFs separated by two intercistronic regions each containing an IRES [62,63]. The two identified IRESs mediate the translation of both the second and third ORFs that encode the MYCHEX1 and c-myc1/c-myc2 proteins, respectively [63].
3. IRES-Dependent Translation Dysregulation-Related Diseases
3.1. Neurodegenerative Diseases
3.1.1. Spinocerebellar Ataxia Type 6 (SCA6)
3.1.2. Fragile X Syndrome
3.1.3. Alzheimer’s Disease
3.1.4. Parkinson’s Disease
3.1.5. Amyotrophic Lateral Sclerosis and Other Neurological Conditions
3.2. Muscular Atrophies
3.2.1. Ischemic Cardiomyopathy (Lymphangiogenesis Regulation)
3.2.2. Myogenesis Regulation
3.2.3. Duchenne Muscular Dystrophy
3.3. Other Specific Diseases
3.3.1. Diamond-Blackfan Anaemia
3.3.2. Diabetes
4. RNA-Based Therapies to Modulate Translation Initiation Dysregulation
4.1. IRESs as Targets
4.2. IRESs as Tools
4.2.1. Parkinson’s Disease
4.2.2. Diabetes
4.2.3. Fabry Disease
4.2.4. Mocupolysaccharidosis III A
4.2.5. Autoimmune Diseases
4.2.6. Cardiovascular Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Barman Balfour, J.A. Fomivirsen: New drug profile. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Krainer, A.R. Antisense-Oligonucleotide Modulation of SMN2 Pre-mRNA Splicing. In Spinal Muscular Atrophy Disease Mechanisms and Therapy; Academic Press: Cambridge, MA, USA, 2017; pp. 301–311. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacerda, R.; Menezes, J.; Romão, L. More than just scanning: The importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell. Mol. Life Sci. 2017, 74, 1659–1680. [Google Scholar] [CrossRef] [PubMed]
- Godet, A.C.; David, F.; Hantelys, F.; Tatin, F.; Lacazette, E.; Garmy-Susini, B.; Prats, A.C. IRES trans-acting factors, key actors of the stress response. Int. J. Mol. Sci. 2019, 20, 924. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Shirokikh, N.E.; Preiss, T. Translation initiation by cap-dependent ribosome recruitment: Recent insights and open questions. Wiley Interdiscip. Rev. RNA 2018, 9, e1473. [Google Scholar] [CrossRef] [PubMed]
- Shatsky, I.N.; Terenin, I.M.; Smirnova, V.V.; Andreev, D.E. Cap-independent translation: What’s in a name? Trends Biochem. Sci. 2018, 43, 882–895. [Google Scholar] [CrossRef]
- Lacerda, R.; Menezes, J.; Candeias, M.M. Alternative Mechanisms of mRNA Translation Initiation in Cellular Stress Response and Cancer. In Advances in Experimental Medicine and Biology; Romão, L., Ed.; Springer: New York, NY, USA, 2019; Volume 1157, pp. 117–132. [Google Scholar]
- Terenin, I.M.; Smirnova, V.V.; Andreev, D.E.; Dmitriev, S.E.; Shatsky, I.N. A researcher’s guide to the galaxy of IRESs. Cell. Mol. Life Sci. 2017, 74, 1431–1455. [Google Scholar] [CrossRef]
- Lackner, D.H.; Bähler, J. Translational control of gene expression: From transcripts to transcriptomes. Int. Rev. Cell Mol. Biol. 2008, 271, 199–251. [Google Scholar] [CrossRef] [PubMed]
- Faye, M.D.; Holcik, M. The role of IRES trans-acting factors in carcinogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Sriram, A.; Bohlen, J.; Teleman, A.A. Translation acrobatics: How cancer cells exploit alternate modes of translational initiation. EMBO Rep. 2018, 19, e45947. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Meng, Z.; Choi, H.; Grizzle, W.E.; Zinn, K.R.; Blume, S.W. Small molecule inhibitors of IRES-mediated translation. Cancer Biol. Ther. 2015, 16, 1471–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, P.; Yu, J.; Ward, R.; Liu, Y.; Hao, Q.; An, S.; Xu, T. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun. Signal 2020, 18, 175. [Google Scholar] [CrossRef] [PubMed]
- Andreev, D.E.; O’Connor, P.B.F.; Loughran, G.; Dmitriev, S.E.; Baranov, P.V.; Shatsky, I.N. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 2017, 45, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haimov, O.; Sinvani, H.; Dikstein, R. Cap-dependent, scanning-free translation initiation mechanisms. Biochim. Biophys. Acta 2015, 1849, 1313–1318. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Molecular Mechanism of Scanning and Start Codon Selection in Eukaryotes. Microbiol. Mol. Biol. Rev. 2011, 75, 434–467. [Google Scholar] [CrossRef] [Green Version]
- Jennings, M.D.; Kershaw, C.J.; Adomavicius, T.; Pavitt, G.D. Fail-safe control of translation initiation by dissociation of eIF2α phosphorylated ternary complexes. eLife 2017, 6, e24542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-Ramos, A.; Candeias, M.M.; Menezes, J.; Lacerda, R.; Willcocks, M.; Teixeira, A.; Locker, N.; Romão, L. Cap-independent translation ensures mTOR expression and function upon protein synthesis inhibition. RNA 2017, 23, 1712–1728. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.; Sonenberg, N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef] [Green Version]
- Balvay, L.; Soto Rifo, R.; Ricci, E.P.; Decimo, D.; Ohlmann, T. Structural and functional diversity of viral IRESes. Biochim. Biophys. Acta 2009, 1789, 542–557. [Google Scholar] [CrossRef]
- Gritsenko, A.A.; Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; de Ridder, D.; Segal, E. Sequence features of viral and human Internal Ribosome Entry Sites predictive of their activity. PLoS Comput. Biol. 2017, 13, e1005734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokrejš, M.; Mašek, T.; Vopálenskỳ, V.; Hlubuček, P.; Delbos, P.; Pospíšek, M. IRESite—A tool for the examination of viral and cellular internal ribosome entry sites. Nucleic Acids Res. 2010, 38, D131. [Google Scholar] [CrossRef] [Green Version]
- Kwan, T.; Thompson, S.R. Noncanonical Translation Initiation in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032672. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, B.; Harris, B.R.E.; Liu, Y.; Deng, Y.; Gradilone, S.A.; Cleary, M.P.; Liu, J.; Yang, D.Q. Targeting IRES-mediated p53 synthesis for cancer diagnosis and therapeutics. Int. J. Mol. Sci. 2017, 18, 93. [Google Scholar] [CrossRef] [Green Version]
- Yaman, I.; Fernandez, J.; Liu, H.; Caprara, M.; Komar, A.A.; Koromilas, A.E.; Zhou, L.; Snider, M.D.; Scheuner, D.; Kaufman, R.J.; et al. The zipper model of translational control: A small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 2003, 113, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.; Yaman, I.; Huang, C.; Liu, H.; Lopez, A.B.; Komar, A.A.; Caprara, M.G.; Merrick, W.C.; Snider, M.D.; Kaufman, R.J.; et al. Ribosome Stalling Regulates IRES-Mediated Translation in Eukaryotes, a Parallel to Prokaryotic Attenuation. Mol. Cell 2005, 17, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Bastide, A.; Karaa, Z.; Bornes, S.; Hieblot, C.; Lacazette, E.; Prats, H.; Touriol, C. An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform. Nucleic Acids Res. 2008, 36, 2434–2445. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.-M.; Shih, Y.-H.; Tseng, J.T.; Lai, M.-C.; Wu, C.-H.; Li, Y.-H.; Tsai, S.-J.; Sun, H.S. Overexpression of FGF9 in colon cancer cells is mediated by hypoxia-induced translational activation. Nucleic Acids Res. 2014, 42, 2932–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-K.; Cheng, R.; Demeter, J.; Chen, J.; Weingarten-Gabbay, S.; Jiang, L.; Snyder, M.P.; Weissman, J.S.; Segal, E.; Jackson, P.K.; et al. Structured elements drive extensive circular RNA translation. Mol. Cell 2021, 81, 4300–4318. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jia, X.; Xu, J. The new function of circRNA: Translation. Clin. Transl. Oncol. Off. Publ. Fed. Spanish Oncol. Soc. Natl. Cancer Inst. Mex. 2020, 22, 2162–2169. [Google Scholar] [CrossRef]
- Kong, S.; Tao, M.; Shen, X.; Ju, S. Translatable circRNAs and lncRNAs: Driving mechanisms and functions of their translation products. Cancer Lett. 2020, 483, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zheng, G.; Ning, Q.; Zheng, J.; Dong, D. Translation and functional roles of circular RNAs in human cancer. Mol. Cancer 2020, 19, 30. [Google Scholar] [CrossRef]
- Tang, X.; Ren, H.; Guo, M.; Qian, J.; Yang, Y.; Gu, C. Review on circular RNAs and new insights into their roles in cancer. Comput. Struct. Biotechnol. J. 2021, 19, 910–928. [Google Scholar] [CrossRef]
- Chen, Y.G.; Chen, R.; Ahmad, S.; Verma, R.; Kasturi, S.P.; Amaya, L.; Broughton, J.P.; Kim, J.; Cadena, C.; Pulendran, B.; et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 2019, 76, 96–109.e9. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, T.; She, Y.; Wu, K.; Gu, S.; Li, L.; Dong, C.; Chen, C.; Zhou, Y. N6-methyladenosine-modified circIGF2BP3 inhibits CD8+ T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol. Cancer 2021, 20, 105. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Karginov, T.A.; Pastor, D.P.H.; Semler, B.L.; Gomez, C.M. Mammalian Polycistronic mRNAs and Disease. Trends Genet. 2017, 33, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surget, S.; Khoury, M.P.; Bourdon, J.C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco. Targets. Ther. 2013, 7, 57–67. [Google Scholar] [PubMed] [Green Version]
- Vieler, M.; Sanyal, S. P53 isoforms and their implications in cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharathchandra, A.; Katoch, A.; Das, S. IRES mediated translational regulation of p53 isoforms. Wiley Interdiscip. Rev. RNA 2013, 5, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Grover, R.; Candeias, M.M.; Fhraeus, R.; Das, S. P53 and little brother p53/47: Linking IRES activities with protein functions. Oncogene 2009, 28, 2766–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaud, E.; Touriol, C.; Boutonnet, C.; Gensac, M.-C.; Vagner, S.; Prats, H.; Prats, A.-C. A New 34-Kilodalton Isoform of Human Fibroblast Growth Factor 2 Is Cap Dependently Synthesized by Using a Non-AUG Start Codon and Behaves as a Survival Factor. Mol. Cell. Biol. 1999, 19, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Huez, I.; Bornes, S.; Bresson, D.; Créancier, L.; Pratz, H. New Vascular Endothelial Growth Factor Isoform Generated by Internal Ribosome Entry Site-Driven CUG Translation Initiation. Mol. Endocrinol. 2001, 15, 2197–2210. [Google Scholar] [CrossRef]
- Huez, I.; Créancier, L.; Audigier, S.; Gensac, M.-C.; Prats, A.-C.; Prats, H. Two Independent Internal Ribosome Entry Sites Are Involved in Translation Initiation of Vascular Endothelial Growth Factor mRNA. Mol. Cell. Biol. 1998, 18, 6178–6190. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.; Kumar, K.N.; Mach, J.R.; Srinivasan, A.; Pal, R.; Bao, X.; Agbas, A.; Höfner, G.; Wanner, K.T.; Michaelis, E.K. A rat brain bicistronic gene with an internal ribosome entry site codes for a phencyclidine-binding protein with cytotoxic activity. J. Biol. Chem. 2009, 284, 2245–2257. [Google Scholar] [CrossRef] [Green Version]
- Secor, J.D.; Fligor, S.C. Free Fatty Acid Receptors as Mediators and Therapeutic Targets in Liver Disease. Front. Physiol. 2021, 12, 656441. [Google Scholar] [CrossRef]
- Halpern, K.B.; Veprik, A.; Rubins, N.; Naaman, O.; Walker, M.D. GPR41 Gene Expression Is Mediated by Internal Ribosome Entry Site (IRES)—Dependent Translation of Bicistronic mRNA. J. Biol. Chem. 2012, 287, 20154–20163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonnelle, D.; Vignard, V.; Sehedic, D.; Moreau-Aubry, A.; Florenceau, L.; Charpentier, M.; Mikulits, W.; Labarriere, N.; Lang, F. The Melanoma Antigens MELOE-1 and MELOE-2 Are Translated from a Bona Fide Polycistronic mRNA Containing Functional IRES Sequences. PLoS ONE 2013, 8, e75233. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, M.; Croyal, M.; Carbonnelle, D.; Fortun, A.; Florenceau, L.; Rabu, C.; Krempf, M.; Labarrière, N.; Lang, F. IRES-dependent translation of the long non coding RNA meloe in melanoma cells produces the most immunogenic MELOE antigens. Oncotarget 2016, 7, 59704–59713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, S.; Bruynooghe, Y.; Denecker, G.; van Huffel, S.; Tinton, S.; Beyaert, R. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol. Cell 2000, 5, 597–605. [Google Scholar] [CrossRef]
- Du, X.; Wang, J.; Zhu, H.; Rinaldo, L.; Lamar, K.-M.; Palmenberg, A.C.; Hansel, C.; Gomez, C.M. Second Cistron in CACNA1A Gene Encodes a Transcription Factor Mediating Cerebellar Development and SCA6. Cell 2013, 154, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Liu, E.; Yan, Y.; Silver, R.T.; Yang, F.; Chen, I.H.; Chen, Y.; Verstovsek, S.; Wang, H.; Prchal, J.; et al. An Unconventional Antigen Translated by a Novel Internal Ribosome Entry Site Elicits Antitumor Humoral Immune Reactions. J. Immunol. 2006, 177, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, A.S.; Overbaugh, J. Evidence that an IRES within the Notch2 coding region can direct expression of a nuclear form of the protein. Mol. Cell 2000, 6, 939–945. [Google Scholar] [CrossRef]
- Anido, J.; Scaltriti, M.; Josep, J.; Serra, B.; Todo, F.R. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006, 25, 3234–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanbru, C.; Lafon, I.; Audigier, S.; Gensac, M.C.; Vagner, S.; Huez, G.; Prats, A.C. Alternative Translation of the Proto-oncogene c-mycby an Internal Ribosome Entry Site. J. Biol. Chem. 1997, 272, 32061. [Google Scholar] [CrossRef] [Green Version]
- Nanbru, C.; Prats, A.-C.; Droogmans, L.; Defrance, P.; Huez, G.; Kruys, V. Translation of the human c-myc P0 tricistronic mRNA involves two independent internal ribosome entry sites. Oncogene 2001, 20, 4270–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, P.D.H.; Du, X.; Fazal, S.; Davies, A.N.; Gomez, C.M. Targeting the CACNA1A IRES as a treatment for spinocerebellar ataxia type 6. PMC 2019, 17, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Du, X.; Muramatsu, S.; Gomez, C.M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. PMC 2017, 8, 347ra94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Gomez, C.M. Spinocerebellar Ataxia Type 6: Molecular Mechanisms and Calcium Channel Genetics. In Polyglutamine Disorders; Nóbrega, C., de Almeida, L., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 147–173. ISBN 978-3-319-71779-1. [Google Scholar]
- Sullivan, R.; Yau, W.Y.; O’Connor, E.; Houlden, H. Spinocerebellar ataxia: An update. J. Neurol. 2019, 266, 533–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.; Riley, K. Fragile X Syndrome. In Encyclopedia of Infant and Early Childhood Development, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 647–654. [Google Scholar] [CrossRef]
- Chen, E.; Sharma, M.R.; Shi, X.; Agrawal, R.K.; Joseph, S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 2014, 54, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-H.; Kim, S.-H.; Jeong, Y.-H.; Kim, S.W.; Min, K.-T.; Kim, K.-T. hnRNP Q regulates internal ribosome entry site-mediated fmr1 translation in neurons. Mol. Cell. Biol. 2018, 39, e00371-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Kim, W.; Lee, K.H.; Kim, S.H.; Lee, H.R.; Kim, H.J.; Jung, Y.; Choi, J.H.; Kim, K.T. HnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ. 2013, 20, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Zhou, Y.; Li, Y.; Han, Y.; Xu, J.; Niu, W.; Li, Z.; Liu, S.; Feng, H.; Huang, W.; et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 2021, 24, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Maloney, B.; Rogers, J.T.; Lahiri, D.K. Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2019, 24, 345–363. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Pehar, M.; Liu, Y.; Bhattacharyya, A.; Zhang, S.C.; O’Riordan, K.J.; Burger, C.; D’Adamio, L.; Puglielli, L. The amyloid precursor protein (APP) intracellular domain regulates translation of p44, a short isoform of p53, through an IRES-dependent mechanism. Neurobiol. Aging 2015, 36, 2725–2736. [Google Scholar] [CrossRef] [Green Version]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. P44, the “longevity-assurance” isoform of P53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef]
- Zhao, F.; Xu, Y.; Gao, S.; Qin, L.; Austria, Q.; Siedlak, S.L.; Pajdzik, K.; Dai, Q.; He, C.; Wang, W.; et al. METTL3-dependent RNA m6A dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol. Neurodegener. 2021, 16, 70. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Plass, M. RNA Dynamics in Alzheimer’s Disease. Molecules 2021, 26, 5113. [Google Scholar] [CrossRef] [PubMed]
- Akhter, R. Circular RNA and Alzheimer’s Disease. Adv. Exp. Med. Biol. 2018, 1087, 239–243. [Google Scholar] [CrossRef]
- Kaluz, S.; Kaluzová, M.; Stanbridge, E.J. Regulation of gene expression by hypoxia: Integration of the HIF-transduced hypoxic signal at the hypoxia-responsive element. Clin. Chim. Acta 2008, 395, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wadlington, N.L.; Chen, L.; Zhuang, X.; Brorson, J.R.; Kang, U.J. Loss of PINK1 attenuates HIF-1α induction by preventing 4E-BP1-dependent switch in protein translation under hypoxia. J. Neurosci. 2014, 34, 3079–3089. [Google Scholar] [CrossRef] [Green Version]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. α-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Bampton, A.; Gittings, L.M.; Fratta, P.; Lashley, T.; Gatt, A. The role of hnRNPs in frontotemporal dementia and amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 140, 599–623. [Google Scholar] [CrossRef]
- Clarke, J.P.; Thibault, P.A.; Salapa, H.E.; Levin, M.C. A Comprehensive Analysis of the Role of hnRNP A1 Function and Dysfunction in the Pathogenesis of Neurodegenerative Disease. Front. Mol. Biosci. 2021, 8, 659610. [Google Scholar] [CrossRef]
- Chen, C.; Ding, X.; Akram, N.; Xue, S.; Luo, S.-Z. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules 2019, 24, 1622. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Wang, H.; Cai, Y.; Wen, H.; Wang, L.; Zhu, M.; Chen, Y.; Yu, Y.; Lu, X.; Zhou, M.; et al. FUS P525L mutation causing amyotrophic lateral sclerosis and movement disorders. Brain Behav. 2020, 10, e01625. [Google Scholar] [CrossRef] [Green Version]
- Morfoisse, F.; Renaud, E.; Hantelys, F.; Prats, A.C.; Garmy-Susini, B. Role of hypoxia and vascular endothelial growth factors in lymphangiogenesis. Mol. Cell. Oncol. 2014, 1, e29907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hantelys, F.; Godet, A.; David, F.; Tatin, F.; Renaud-Gabardos, E.; Pujol, F.; Diallo, L.; Ader, I.; Ligat, L.; Anthony, K.; et al. Vasohibin1, a new IRES trans-acting factor for induction of (lymph)angiogenic factors in early hypoxia. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Aspriţoiu, V.M.; Stoica, I.; Bleotu, C.; Diaconu, C.C. Epigenetic Regulation of Angiogenesis in Development and Tumors Progression: Potential Implications for Cancer Treatment. Front. Cell Dev. Biol. 2021, 9, 2462. [Google Scholar] [CrossRef] [PubMed]
- Morfoisse, F.; Kuchnio, A.; Frainay, C.; Gomez-Brouchet, A.; Delisle, M.-B.; Marzi, S.; Helfer, A.-C.; Hantelys, F.; Pujol, F.; Guillermet-Guibert, J.; et al. Hypoxia Induces VEGF-C Expression in Metastatic Tumor Cells via a HIF-1α-Independent Translation-Mediated Mechanism. Cell Rep. 2014, 6, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Conte, C.; Ainaoui, N.; Delluc-Claviéres, A.; Khoury, M.P.; Azar, R.; Pujol, F.; Martineau, Y.; Pyronnet, S.; Prats, A.C. Fibroblast growth factor 1 induced during myogenesis by a transcription-translation coupling mechanism. Nucleic Acids Res. 2009, 37, 5267–5278. [Google Scholar] [CrossRef] [Green Version]
- Ainaoui, N.; Hantelys, F.; Renaud-Gabardos, E.; Bunel, M.; Lopez, F.; Pujol, F.; Planes, R.; Bahraoui, E.; Pichereaux, C.; Burlet-Schiltz, O.; et al. Promoter-dependent translation controlled by p54nrb and hnRNPM during myoblast differentiation. PLoS ONE 2015, 10, e0136466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Chao, Z.; Zhang, R.; Ding, R.; Wang, Y.; Wu, W.; Han, Q.; Li, C.; Xu, H.; Wang, L.; et al. Circular RNA Regulation of Myogenesis. Cells 2019, 8, 885. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, M.; Lian, D.; Li, Y.; Li, Y.; Wang, J.; Deng, S.; Yu, K.; Lian, Z. Non-Coding RNA Regulates the Myogenesis of Skeletal Muscle Satellite Cells, Injury Repair and Diseases. Cells 2019, 8, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchenne Muscular Dystrophy—NORD (National Organization for Rare Disorders). Available online: https://rarediseases.org/rare-diseases/duchenne-muscular-dystrophy/ (accessed on 21 June 2022).
- Abreu, N.J.; Waldrop, M.A. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr. Pulmonol. 2021, 56, 710–720. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef]
- Blake, D.J.; Tinsley, J.M.; Davies, K.E. Utrophin: A structural and functional comparison to dystrophin. Brain Pathol. 1996, 6, 37–47. [Google Scholar] [CrossRef]
- Miura, P.; Coriati, A.; Bélanger, G.; de Repentigny, Y.; Lee, J.; Kothary, R.; Holcik, M.; Jasmin, B.J. The utrophin A 5′-UTR drives cap-independent translation exclusively in skeletal muscles of transgenic mice and interacts with eEF1A2. Hum. Mol. Genet. 2010, 19, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, P.; Andrews, M.; Holcik, M.; Jasmin, B.J. IRES-mediated translation of utrophin A is enhanced by glucocorticoid treatment in skeletal muscle cells. PLoS ONE. 2008, 3, e2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, P.; Thompson, J.; Chakkalakal, J.V.; Holcik, M.; Jasmin, B.J. The utrophin A 5′-untranslated region confers internal ribosome entry site-mediated translational control during regeneration of skeletal muscle fibers. J. Biol. Chem. 2005, 280, 32997–33005. [Google Scholar] [CrossRef] [Green Version]
- Soblechero-Martín, P.; López-Martínez, A.; de la Puente-Ovejero, L.; Vallejo-Illarramendi, A.; Arechavala-Gomeza, V. Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol. Appl. Neurobiol. 2021, 47, 711–723. [Google Scholar] [CrossRef]
- Engidaye, G.; Melku, M.; Enawgaw, B. Diamond Blackfan Anemia: Genetics, Pathogenesis, Diagnosis and Treatment. EJIFCC 2019, 30, 67–81. [Google Scholar] [PubMed]
- Horos, R.; IJspeert, H.; Pospisilova, D.; Sendtner, R.; Andrieu-Soler, C.; Taskesen, E.; Nieradka, A.; Cmejla, R.; Sendtner, M.; Touw, I.P.; et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 2012, 119, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, K.A.; Cobbold, L.C.; Ridley, S.H.; Coldwell, M.; Bottley, A.; Bushell, M.; Willis, A.E.; Siddle, K. The human insulin receptor mRNA contains a functional internal ribosome entry segment. Nucleic Acids Res. 2009, 37, 5881–5893. [Google Scholar] [CrossRef] [Green Version]
- Marr, M.T.; D’Alessio, J.A.; Puig, O.; Tjian, R. IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev. 2007, 21, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobbold, L.C.; Spriggs, K.A.; Haines, S.J.; Dobbyn, H.C.; Hayes, C.; de Moor, C.H.; Lilley, K.S.; Bushell, M.; Willis, A.E. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol. Cell. Biol. 2008, 28, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Péladeau, C.; Jasmin, B.J. Targeting IRES-dependent translation as a novel approach for treating Duchenne muscular dystrophy. RNA Biol. 2020, 18, 1238–1251. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K. RNA therapy: Rich history, various applications and unlimited future prospects. Exp. Mol. Med. 2022, 54, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Choi, Y.H.; Tu, M.J. RNA drugs and RNA targets for small molecules: Principles, progress, and challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Exploring internal ribosome entry sites as therapeutic targets. Front. Oncol. 2015, 5, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Karaki, S. Antisense Oligonucleotides, A Novel Developing Targeting Therapy. In Antisense Therapy; Paris, C., Ed.; IntechOpen: Rijeka, Croatia, 2019; p. 10. ISBN 978-1-78984-533-4. [Google Scholar]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense oligonucleotide: Basic concepts and therapeutic application in inflammatory bowel disease. Front. Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef] [Green Version]
- Chery, J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.-Y.; Qin, Z.; Zhu, Y.-H.; He, Z.-Y.; Xu, T. Current RNA-based Therapeutics in Clinical Trials. Curr. Gene Ther. 2019, 19, 172–196. [Google Scholar] [CrossRef] [PubMed]
- Guerniou, V.; Gillet, R.; Berrée, F.; Carboni, B.; Felden, B. Targeted inhibition of the hepatitis C internal ribosomal entry site genomic RNA with oligonucleotide conjugates. Nucleic Acids Res. 2007, 35, 6778–6787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallet-Lopez, B.; Aldaz-Carroll, L.; Chabas, S.; Dausse, E.; Staedel, C.; Toulme, J.J. Antisense oligonucleotides targeted to the domain IIId of the hepatitis C virus IRES compete with 40S ribosomal subunit binding and prevent in vitro translation. Nucleic Acids Res. 2003, 31, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Sun, H.; Shen, W.; Wang, S.; Yao, J.; Migawa, M.T.; Bui, H.H.; Damle, S.S.; Riney, S.; Graham, M.J.; et al. Antisense oligonucleotides targeting translation inhibitory elements in 5′ UTRs can selectively increase protein levels. Nucleic Acids Res. 2017, 45, 9528–9546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahel, R.A.; Zangemeister-Wittke, U. Antisense oligonucleotides for cancer therapy—An overview. Lung Cancer 2003, 41, 81–88. [Google Scholar] [CrossRef]
- Holcik, M. Targeting Translation for Treatment of Cancer—A Novel Role for IRES? Curr. Cancer Drug Targets 2004, 4, 299–311. [Google Scholar] [CrossRef]
- Tarn, W.; Cheng, Y.; Ko, S.; Huang, L. Antisense Oligonucleotide-Based Therapy of Viral Infections. Pharmaceutics 2021, 13, 2015. [Google Scholar] [CrossRef]
- Park, E.; Maquat, L.E. Staufen-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 2013, 4, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcellus, K.A.; Crawford Parks, T.E.; Almasi, S.; Jasmin, B.J. Distinct roles for the RNA-binding protein Staufen1 in prostate cancer. BMC Cancer 2021, 21, 120. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.; Monette, A.; Niu, M.; Barrera, A.; López-Ulloa, B.; Fuentes, Y.; Guizar, P.; Pino, K.; DesGroseillers, L.; Mouland, A.J.; et al. The double-stranded RNA-binding protein, Staufen1, is an IRES-transacting factor regulating HIV-1 cap-independent translation initiation. Nucleic Acids Res. 2022, 50, 411–429. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA vaccine era—Mechanisms, drug platform and clinical prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didiot, M.C.; Hewett, J.; Varin, T.; Freuler, F.; Selinger, D.; Nick, H.; Reinhardt, J.; Buckler, A.; Myer, V.; Schuffenhauer, A.; et al. Identification of cardiac glycoside molecules as inhibitors of c-Myc IRES-mediated translation. J. Biomol. Screen. 2013, 18, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.V.; King, L.Y.; Chung, R.T. Hepatitis C Virus-Associated Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 345–370. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tahara, H.; Cai, Q.; Storkus, W.J.; Muller, G.; Maurice, G.; Wolf, S.F.; Robbins, P.D.; Lotze, M.T. Construction and characterization of retroviral vectors expressing biologically active human interleukin-12. Hum. Gene Ther. 1994, 5, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Scappaticci, F.A.; Smith, R.; Pathak, A.; Schloss, D.; Lum, B.; Cao, Y.; Johnson, F.; Engleman, E.G.; Nolan, G.P. Combination Angiostatin and Endostatin Gene Transfer Induces Synergistic Antiangiogenic Activity in Vitro and Antitumor Efficacy in Leukemia and Solid Tumors in Mice. Mol. Ther. 2001, 3, 186–196. [Google Scholar] [CrossRef]
- Ohlfest, J.R.; Demorest, Z.L.; Motooka, Y.; Vengco, I.; Oh, S.; Chen, E.; Scappaticci, F.A.; Saplis, R.J.; Ekker, S.C.; Low, W.C.; et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the sleeping beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Mol. Ther. 2005, 12, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Kupatt, C.; Hinkel, R.; Pfosser, A.; El-Aouni, C.; Wuchrer, A.; Fritz, A.; Globisch, F.; Thormann, M.; Horstkotte, J.; Lebherz, C.; et al. Cotransfection of vascular endothelial growth factor-A and platelet-derived growth factor-B via recombinant adeno-associated virus resolves chronic ischemic malperfusion role of vessel maturation. J. Am. Coll. Cardiol. 2010, 56, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukuła, K.; Chojnowska, L.; Dąbrowski, M.; Witkowski, A.; Chmielak, Z.; Skwarek, M.; Kądziela, J.; Teresińska, A.; Małecki, M.; Janik, P.; et al. Intramyocardial plasmid-encoding human vascular endothelial growth factor A165/basic fibroblast growth factor therapy using percutaneous transcatheter approach in patients with refractory coronary artery disease (VIF-CAD). Am. Heart J. 2011, 161, 581–589. [Google Scholar] [CrossRef]
- Douin, V.; Bornes, S.; Creancier, L.; Rochaix, P.; Favre, G.; Prats, A.-C.; Couderc, B. Use and comparison of different internal ribosomal entry sites (IRES) in tricistronic retroviral vectors. BMC Biotechnol. 2004, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villaflores, O.B.; Chen, Y.J.; Chen, C.P.; Yeh, J.M.; Wu, T.Y. Effects of curcumin and demethoxycurcumin on amyloid-β precursor and tau proteins through the internal ribosome entry sites: A potential therapeutic for Alzheimer’s disease. Taiwan. J. Obstet. Gynecol. 2012, 51, 554–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Allaf, F.A.; Abduljaleel, Z.; Athar, M.; Taher, M.M.; Khan, W.; Mehmet, H.; Colakogullari, M.; Apostolidou, S.; Bigger, B.; Waddington, S.; et al. Modifying inter-cistronic sequence significantly enhances IRES dependent second gene expression in bicistronic vector: Construction of optimised cassette for gene therapy of familial hypercholesterolemia. Non-Coding RNA Res. 2019, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Renaud-Gabardos, E.; Hantelys, F.; Morfoisse, F.; Chaufour, X.; Garmy-Susini, B.; Prats, A.-C. Internal ribosome entry site-based vectors for combined gene therapy. World J. Exp. Med. 2015, 5, 11–20. [Google Scholar] [CrossRef]
- Zangi, L.; Lui, K.O.; von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Später, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R.; et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-L.; Leblond, A.-L.; Turner, E.C.; Kumar, A.H.; Martin, K.; Whelan, D.; O’Sullivan, D.M.; Caplice, N.M. Synthetic Chemically Modified mRNA-Based Delivery of Cytoprotective Factor Promotes Early Cardiomyocyte Survival Post-Acute Myocardial Infarction. Mol. Pharm. 2015, 12, 991–996. [Google Scholar] [CrossRef]
- Lu, D.; Thum, T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Abdrakhmanova, I.I.; Chernov, V.M.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Production and application of multicistronic constructs for various human disease therapies. Pharmaceutics 2019, 11, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzouz, M.; Martin-Rendon, E.; Barber, R.D.; Mitrophanous, K.A.; Carter, E.E.; Rohll, J.B.; Kingsman, S.M.; Kingsman, A.J.; Mazarakis, N.D. Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 10302–10312. [Google Scholar] [CrossRef] [Green Version]
- Palfi, S.; Gurruchaga, J.M.; Scott Ralph, G.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef]
- Palfi, S.; Gurruchaga, J.M.; Lepetit, H.; Howard, K.; Ralph, G.S.; Mason, S.; Gouello, G.; Domenech, P.; Buttery, P.C.; Hantraye, P.; et al. Long-Term Follow-Up of a Phase I/II Study of ProSavin, a Lentiviral Vector Gene Therapy for Parkinson’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.J.; Reger, R.L.; Alexander, G.M.; Class, R.; Azizi, S.A.; Prockop, D.J. Rat marrow stromal cells rapidly transduced with a self-inactivating retrovirus synthesize L-DOPA in vitro. Gene Ther. 2001, 8, 1214–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Huang, Y.; Guo, Q.; Fan, X.; Lu, Y.; Zhu, S.; Wang, Y.; Bo, X.; Chang, X.; Zhu, M.; et al. Differentiation of iPSCs into insulin-producing cells via adenoviral transfection of PDX-1, NeuroD1 and MafA. Diabetes Res. Clin. Pract. 2014, 104, 383–392. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Aksentijevich, I.; Murray, G.J.; Brady, R.O.; Pastan, I.; Gottesman, M.M. Retroviral coexpression of a multidrug resistance gene (MDR1) and human alpha-galactosidase A for gene therapy of Fabry disease. Hum. Gene Ther. 1995, 6, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zérah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh-Sikaroodi, S.; Hosseini, A.; Fallah, A.; Estiri, H.; Noormohammadi, Z.; Salehi, M.; Ghaderian, S.M.H.; Akhavan Niaki, H.; Soleimani, M.; Kazemi, B. Lentiviral Mediating Genetic Engineered Mesenchymal Stem Cells for Releasing IL-27 as a Gene Therapy Approach for Autoimmune Diseases. Cell J. 2014, 16, 255–262. [Google Scholar]
- Rayssac, A.; Neveu, C.; Pucelle, M.; van den Berghe, L.; Prado-Lourenco, L.; Arnal, J.-F.; Chaufour, X.; Prats, A.-C. IRES-based vector coexpressing FGF2 and Cyr61 provides synergistic and safe therapeutics of lower limb ischemia. Mol. Ther. 2009, 17, 2010–2019. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, K.; Qiang, H.; Tang, Y.; Li, Q.; Li, M.; Dang, X. Angiopoiesis and bone regeneration via co-expression of the hVEGF and hBMP genes from an adeno-associated viral vector in vitro and in vivo. Acta Pharmacol. Sin. 2010, 31, 821–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazwa, A.; Tomczyk, M.; Taha, H.M.; Hytonen, E.; Stoszko, M.; Zentilin, L.; Giacca, M.; Yla-Herttuala, S.; Emanueli, C.; Jozkowicz, A.; et al. Arteriogenic therapy based on simultaneous delivery of VEGF-A and FGF4 genes improves the recovery from acute limb ischemia. Vasc. Cell 2013, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Feng, Y.; Lu, W.; Li, H.; Xie, H.; Li, S. Effect of combined VEGF165/SDF-1 gene therapy on vascular remodeling and blood perfusion in cerebral ischemia. 2016, 127, 670–678. J. Neurosurg. 2016, 127, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif, S.; Jahangeer, M.; Maknoon Razia, D.; Ashiq, M.; Ghaffar, A.; Akram, M.; El Allam, A.; Bouyahya, A.; Garipova, L.; Ali Shariati, M.; et al. Dopamine in Parkinson’s disease. Clin. Chim. Acta. 2021, 522, 114–126. [Google Scholar] [CrossRef]
- Muthuraman, M.; Koirala, N.; Ciolac, D.; Pintea, B.; Glaser, M.; Groppa, S.; Tamás, G.; Groppa, S. Deep Brain Stimulation and L-DOPA Therapy: Concepts of Action and Clinical Applications in Parkinson’s Disease. Front. Neurol. 2018, 9, 711. [Google Scholar] [CrossRef]
- Gillespie, K.M. Type 1 diabetes: Pathogenesis and prevention. Can. Med. Assoc. J. 2006, 175, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Mu, Y.; Hou, L.; Liu, J. Examining the therapeutic potential of various stem cell sources for differentiation into insulin-producing cells to treat diabetes. Ann. Endocrinol. 2019, 80, 47–53. [Google Scholar] [CrossRef]
- Kaneto, H.; Matsuoka, T.; Kawashima, S.; Yamamoto, K.; Kato, K.; Miyatsuka, T.; Katakami, N.; Matsuhisa, M. Role of MafA in pancreatic β-cells. Adv. Drug Deliv. Rev. 2009, 61, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Pacienza, N.; Yoshimitsu, M.; Mizue, N.; Au, B.C.Y.; Wang, J.C.M.; Fan, X.; Takenaka, T.; Medin, J.A. Lentivector transduction improves outcomes over transplantation of human HSCs alone in NOD/SCID/Fabry mice. Mol. Ther. 2012, 20, 1454–1461. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Gregersen, P.K. Genomics and the Multifactorial Nature of Human Autoimmune Disease. N. Engl. J. Med. 2011, 365, 1612–1623. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Pathologies | IRES-Containing Transcripts | Related ITAFs | Tested RNA-Based Therapies | References | |
|---|---|---|---|---|---|
| Neurodegenerative diseases | Spinocerebellar ataxia type 6 | CACNA1A | n.i. * | miRNA-based therapy | [65,66] |
| Fragile X syndrome | fmr1 | hnRNPQ | n.i. * | [70] | |
| Alzheimer’s disease | p53 (p44 isoform) | APP (AICD), nucleolin | n.i. * | [74] | |
| Parkinson’s disease | HIF-1α | PINK1 | Antisense oligo nucleotide reducing the expression of α-synuclein pathogenic protein | [82,83] | |
| Amyotrophic lateral sclerosis | Related RBPs: hnRNPA2/B1, hnRNPA1, FUS | n.i. * | [84] | ||
| Muscular atrophies | Ischemic cardiomyopathy | VEGFA, VEGFC, FGF1 | hnRNPL, VASH1 | n.i. * | [90,91] |
| Myogenesis regulation | FGF1/FGF2 | hnRNPM, p54nrb | n.i. * | [95,110] | |
| Duchenne muscular dystrophy | utrophin A | eEF1A2 | IRES over-expression by small molecules | [102,111] | |
| Other diseases | Diamond-Blackfan anaemia | Bag1/Csde1, p53 | Rps19, Rpl11 | n.i. * | [107] |
| Diabetes | INR/IGF-1R | PTBP1, HuR, hnRNPC | miRNA-based therapy | [108,109] |
| Disease/Condition | IRES Gene Therapy | Expressed Proteins | Purpose | References |
|---|---|---|---|---|
| Parkinson’s disease |
|
|
| [147,148,149,150] |
| Diabetes | Multicistronic adenoviral construct | Pancreatic and duodenal homeobox-1 (Pdx1), Neurogenin 3 (Ngn3) V-musculoaponeurotic fibrosarcoma oncogene homolog A (MafA) | Reprogramming of hepatocytes into insulin-producing cells in vitro and correcting the diabetic state in vivo | [151] |
| Fabry disease | Bicistronic retroviral vectors | a-Gal A gene drug-selectable multidrug resistance gene 1 (MDR1) | Restore the deficiency of the α-galactosidase A (a-Gal A) enzyme | [152] |
| Mucopoly- saccharidosis IIIA | Adeno-associated virus (AAV)-based bicistronic vector | Heparan-N-sulfamidase and N-sulfoglycosamine sulfohydrolase (SGSH) Sulfatase-modifying factor (SUMF1) | Improve heparan sulfate catabolism and decrease microglial activation | [153] |
| Autoimmune diseases | Bicistronic lentiviral vector | Two IL-27 subunits (p28 and EBI3) | Promote the differentiation of T-cells that secrete IL-10 | [154] |
| Cardiovascular diseases |
|
|
| [155,156,157,158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, R.; Lacerda, R.; Romão, L. Internal Ribosome Entry Site (IRES)-Mediated Translation and Its Potential for Novel mRNA-Based Therapy Development. Biomedicines 2022, 10, 1865. https://doi.org/10.3390/biomedicines10081865
Marques R, Lacerda R, Romão L. Internal Ribosome Entry Site (IRES)-Mediated Translation and Its Potential for Novel mRNA-Based Therapy Development. Biomedicines. 2022; 10(8):1865. https://doi.org/10.3390/biomedicines10081865
Chicago/Turabian StyleMarques, Rita, Rafaela Lacerda, and Luísa Romão. 2022. "Internal Ribosome Entry Site (IRES)-Mediated Translation and Its Potential for Novel mRNA-Based Therapy Development" Biomedicines 10, no. 8: 1865. https://doi.org/10.3390/biomedicines10081865
APA StyleMarques, R., Lacerda, R., & Romão, L. (2022). Internal Ribosome Entry Site (IRES)-Mediated Translation and Its Potential for Novel mRNA-Based Therapy Development. Biomedicines, 10(8), 1865. https://doi.org/10.3390/biomedicines10081865