
Direct Oral Anticoagulants (DOACs) for Therapeutic Targeting of Thrombin, a Key Mediator of Cerebrovascular and Neuronal Dysfunction in Alzheimer’s Disease


Abstract
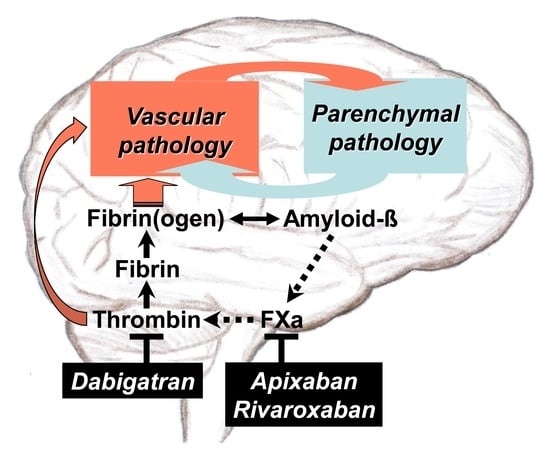
:
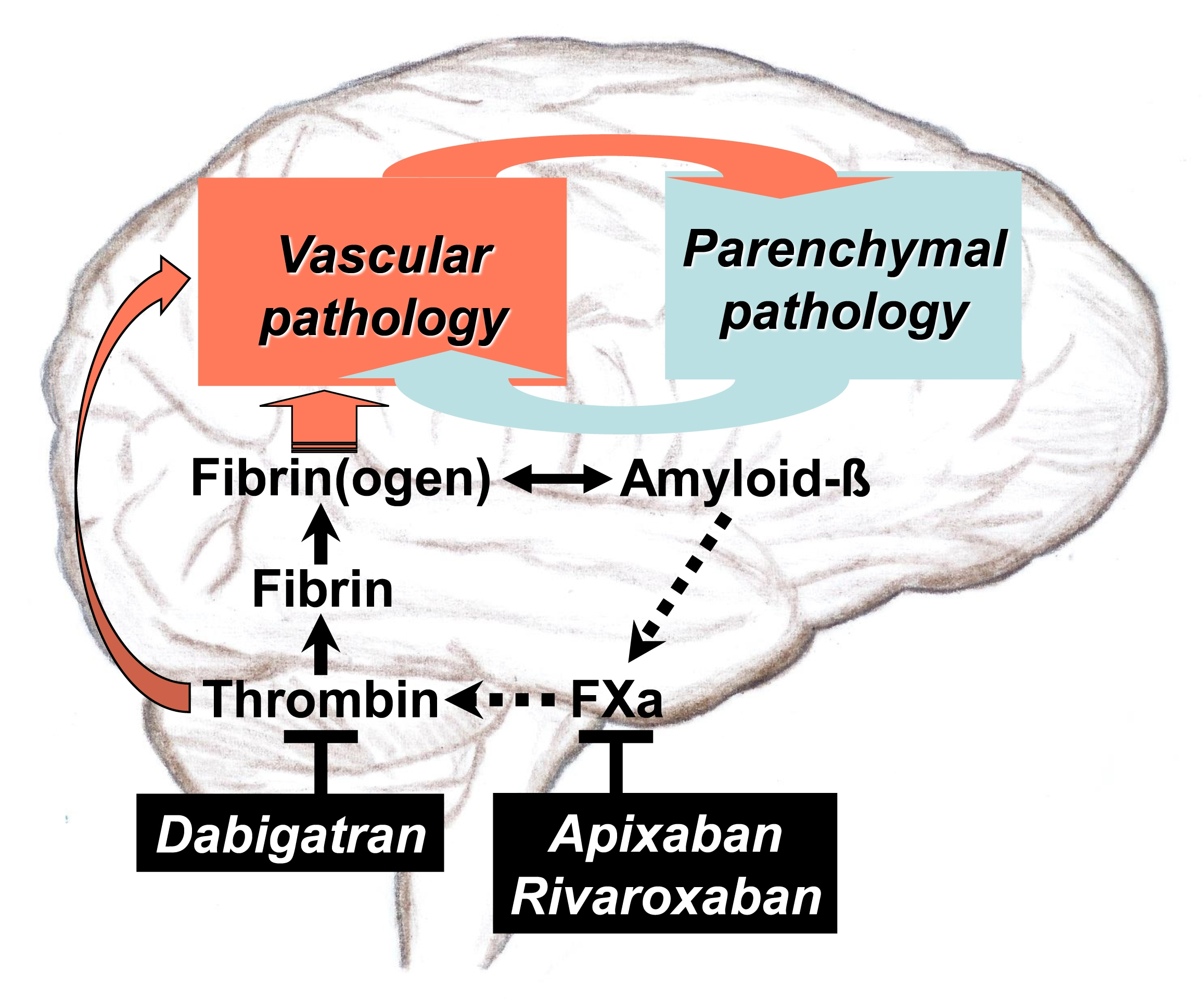{kind=link}
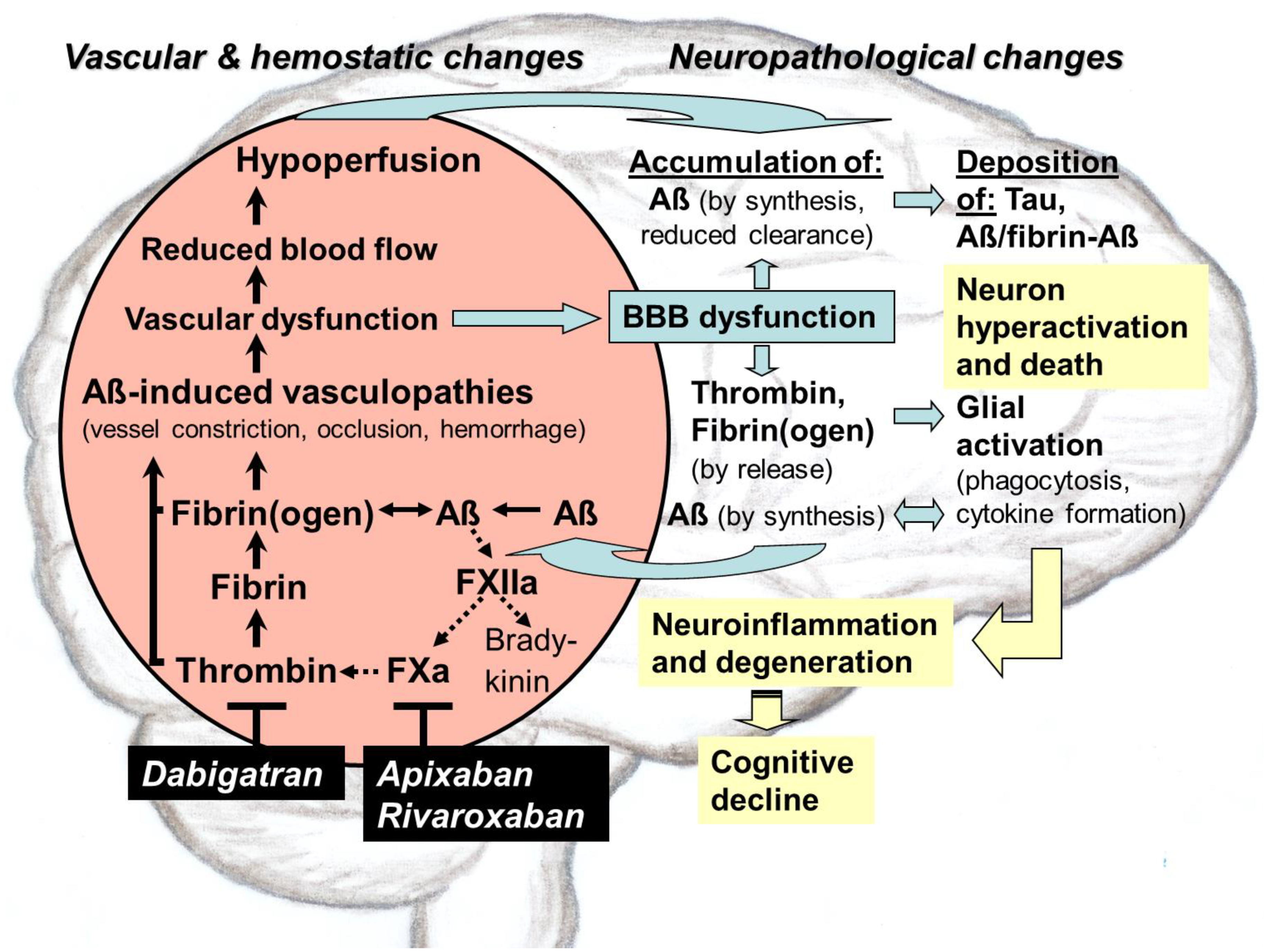
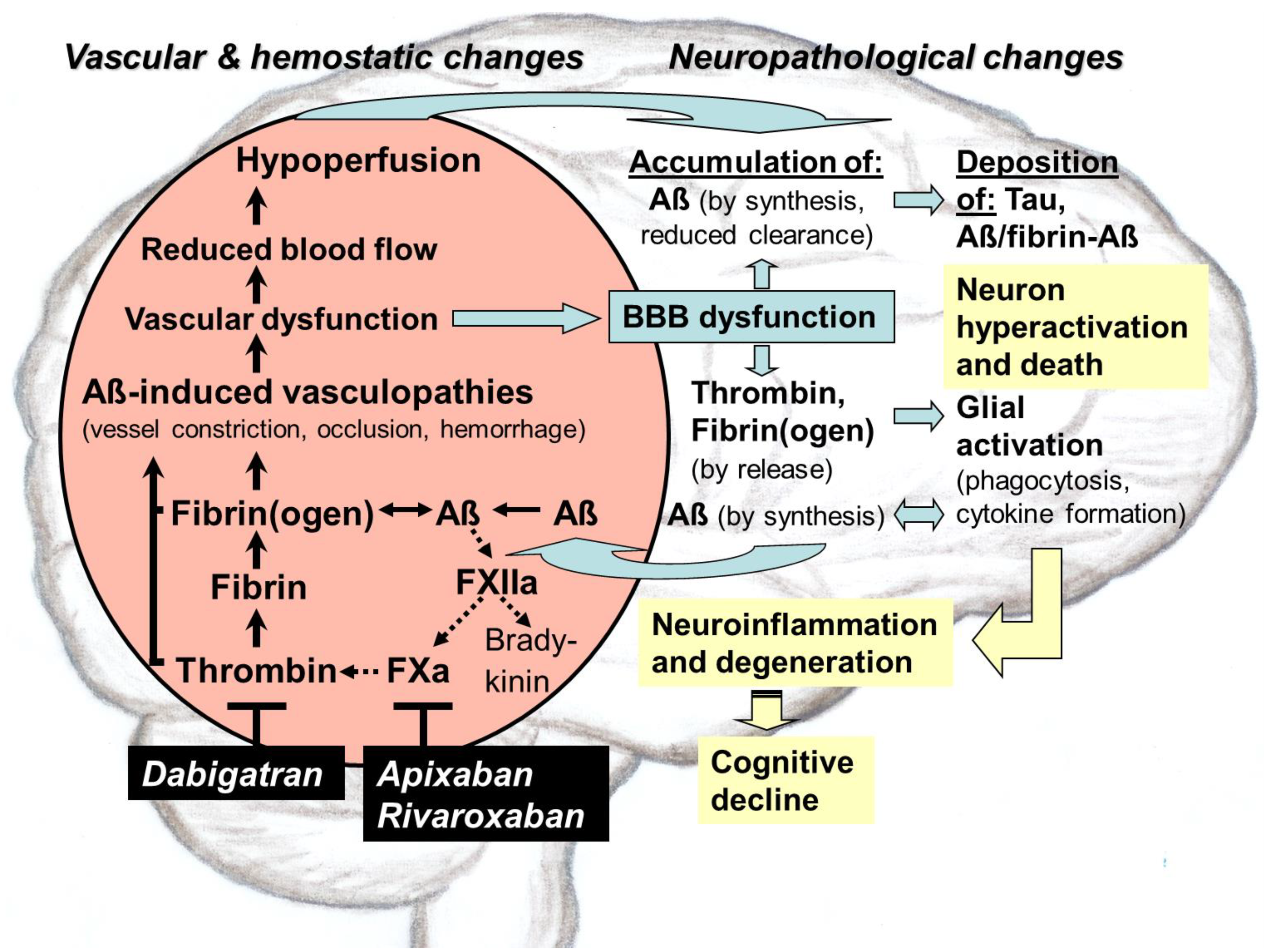{kind=link}
1. Introduction
2. Hemostasis, Thrombosis, and Antithrombotic Medication
2.1. Blood Coagulation and Fibrinolysis
2.2. Antithrombotic Therapy
2.2.1. Drug Portfolio
2.2.2. Fields of Indications
3. Toxic Proteins and Chronic Inflammation in AD
3.1. Generation and Occurrence of Aß
3.2. Brain Locations and Pathogenic Action of Aß
3.2.1. Evidence for Aß Causality
3.2.2. Aß-Targeting Antibody Therapy
3.3. Tau Protein Pathologies
3.4. Inflammation and Glial Responses
3.5. General Remarks
4. Role of Aß in Triggering Vascular Constriction and CAA in AD
4.1. AD Mouse Models
4.2. Occurrence of CAA
4.3. Aß in CAA and Brain Parenchyma
4.4. Brain Vasculopathies and Lesions by Aß-Driven CAA
4.5. Pathophysiological Impact of Aß on Vascular and BBB Functioning
4.5.1. Decrease in CBF Induced by Capillary Constriction and CAA
4.5.2. BBB Dysfunction and Impaired Aß Clearance
4.5.3. Target for Therapeutics
5. Interaction of Aß with the Plasma Contact System and Its Driven Pathways of Coagulation and Inflammation in AD
5.1. Aß-Induced Activation of FXII in Contact System and Effects on Pathways Beyond
5.1.1. Indications for Aß Causality
5.1.2. Aß-Induced Accumulation of Thrombin and Fibrin(ogen)
5.1.3. Formation of Aß-Containing Fibrin Clots
5.2. Pathological Dimension
6. Therapeutical Intervention Using Thrombin-Inhibiting Anticoagulants against Dysregulated Intrinsic Coagulation in AD
6.1. Rationales for Use: Results from Basic Research
6.2. Rationales for Use: Results from Preclinical Studies
6.3. Rationales for Use: Results from Clinical Studies
6.3.1. Historical View on Early Investigations and the Hypothesis of AD Therapy
6.3.2. Observer Studies on Patients with Anticoagulant Use due to AF
7. Clinical Perspective for Anticoagulant Use against AD
7.1. Evaluation of Therapeutic Suitability of Available Anticoagulants
7.1.1. Parenteral Anticoagulants
7.1.2. Oral Anticoagulants
7.1.3. Risk Assessment of Oral Anticoagulants in Clinical Observer Studies
7.2. DOAC-Type Anticoagulants for In-Depth Clinical Investigation
7.2.1. Direct Thrombin Inhibitor Dabigatran
7.2.2. FXa-Inhibitors Apixaban and Rivaroxaban
7.3. Concluding Remarks
8. Conception for a Clinical Intervention Study with DOAC Treatment
8.1. Bleeding Risk and CAA Development
8.2. Methods for AD Diagnosis
8.2.1. Interviews and Neuropsychological Testing
8.2.2. EEG, MRI, and PET Imaging
8.2.3. Invasive CSF Analysis
8.2.4. Diagnosis by Blood-Based Tests and Intestinal Microbiome Analysis
8.3. Clinical Perspective of DOACs
8.3.1. Limitations and Qualification Scenarios for Investigation
8.3.2. Drug Options for Therapeutic Approach
8.3.3. Future Direction towards DOAC Repositioning for AD
8.3.4. Other Brain Amyloidosis with Associated Vascular Dysfunction
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aß | Amyloid-ß proteins |
| AßPP | Amyloid-ß protein precursor |
| AD | Alzheimer´s disease |
| AF | Atrial fibrillation |
| ApoE | Apolipoprotein E |
| ATN | Amyloid, tau, and neurodegeneration |
| BBB | Blood–brain barrier |
| CAA | Aß-type cerebral amyloid angiopathy |
| CBF | Cerebral blood flow |
| cHK | Cleaved HK |
| CSF | Cerebrospinal fluid |
| DOAC | Direct oral anticoagulant |
| EEG | Electroencephalography |
| FDA | U.S. Food and Drug Administration |
| FSH | Follicle-stimulating hormone |
| FXI | Blood coagulation factor XI |
| FXII | Blood coagulation factor XII |
| FXa | Activated factor X |
| FXIIa | Activated factor XII |
| ISF | Interstitial fluid |
| HK | High molecular weight kininogen |
| MRI | Magnetic resonance imaging |
| NfL | Neurofilament light chain protein |
| PAR | Protease-activated receptor |
| PET | Position emission tomography |
| PD | Parkinson´s disease |
| PK | Prekallikrein |
| pTau | Hyperphosphorylated tau protein |
| ROS | Reactive oxygen species |
| t-PA | Tissue plasminogen activator |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| VKA | Vitamin K antagonist |
References
- Bickel, H. Die Häufigkeit von Demenzerkrankungen. Inf. Dtsch. Ges. Selbsthilfe Demenz Berl. 2020, 1, 1–10. [Google Scholar]
- Abbott, A. Treating Alzheimer’s before it takes hold. Nature 2022, 603, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.; Escott-Price, V.; De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 2020, 370, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, D.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Past, present and future of therapeutic strategies against amyloid-ß peptides in Alzheimer’s disease: A systematic review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef]
- Strickland, S. Blood will out: Vascular contributions to Alzheimer’s disease. J. Clin. Investig. 2018, 128, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Chen, Y.; Mao, R.; Wang, J.; Liu, R.; Liu, Y.; Wang, X. Advances in drug therapy for Alzheimer’s disease. Curr. Med. Sci. 2020, 40, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310. [Google Scholar] [CrossRef]
- Bauzon, J.; Lee, G.; Cummings, J. Repurposed agents in the Alzheimer’s disease drug development pipeline. Alzheimer’s Res. Ther. 2020, 13, 98. [Google Scholar] [CrossRef]
- Grossmann, K. Anticoagulants for treatment of Alzheimer’s disease. J. Alzheimers Dis. 2020, 77, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K. Direct oral anticoagulants: A new therapy against Alzheimer’s disease? Neural. Reg. Res. 2021, 16, 1556–1557. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K. Alzheimer’s disease—Rationales for potential treatment with the thrombin inhibitor dabigatran. Int. J. Mol. Sci. 2021, 22, 4805. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, S.; Hermkens, A.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M.J.A.P. Vascular hypothesis of Alzheimer’s disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Badimon, A.; Chen, Z.-L.; Strickland, S.; Norris, E.H. The contact activation system and vascular factors as alternative targets for Alzheimer’s disease therapy. Res. Pract. Thromb. Haemost. 2021, 5, e12504. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K. Alzheimer-Krankheit–können Antikoagulantien helfen? J. Neurol. Neurochir. Psychiat. 2021, 22, 7–10. [Google Scholar]
- Grosser, T.; Weber, A.-A. Pharmakologie der Hämostase. In Allgemeine und spezielle Pharmakologie und Toxikologie, 12th ed.; Aktories, K., Förstermann, U., Hofmann, F., Starke, K., Eds.; Elsevier: München, Germany, 2017; pp. 465–488. [Google Scholar]
- Petersen, M.A.; Ryu, J.K.; Akassoglou, K. Fibrinogen in neurological diseases: Mechanisms, imaging and therapeutics. Nat. Rev. 2018, 19, 283–301. [Google Scholar] [CrossRef]
- Martin, R. Targeting fibrin in neurodegeneration. Nat. Immunol. 2018, 19, 1149–1150. [Google Scholar] [CrossRef] [PubMed]
- Kresge, N.; Simoni, R.D.; Hill, R.L. Hemorrhagic sweet clover disease, dicumarol, and warfarin: The work of Karl Paul Link. J. Biolog. Chem. 2005, 280, e5–e6. [Google Scholar] [CrossRef]
- Shameem, R.; Ansell, J. Disadvantages of VKA and requirements for novel anticoagulants. Best. Pract. Res. Clin. Haematol. 2013, 26, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Klimke, K.; Paschke, L.; Schulz, M. Orale Antikoagulantien. In Rx-Trendbericht: Thema im Fokus; Zentralinstitut für die Kassenärztliche Versorgung in Deutschland: Berlin, Germany, 2019; pp. 1–5. [Google Scholar]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. 1907, 64, 146–148. [Google Scholar]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Alzheimer’s and parkinson’s diseases: The prion concept in relation to assembled Aß, tau, and α-synuclein. Science 2015, 349, 601. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Cerebral amyloid angiopathy: Emerging concepts. J. Stroke 2015, 17, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The physiological roles of amyloid-ß peptide hint at new ways to treat Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.-J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of ß amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef]
- Selkoe, D.J. Treatments for Alzheimer’s disease emerge. Science 2021, 373, 624–626. [Google Scholar] [CrossRef]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lövestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM structures of amyloid-ß 42 filaments from human brain. Science 2022, 375, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, V.; Andre, I.; Gasser, U.; Dubackic, M.; Olsson, U.; Linse, S. Amyloid-ß 42 fibril structure based on small-angle scattering. Proc. Natl. Acad. Sci. USA 2021, 118, e2112783118. [Google Scholar] [CrossRef]
- Ghosh, U.; Yau, W.-M.; Collinge, J.; Tycko, R. Structural differences in amyloid-ß fibrils from brains of nondemented elderly individuals and Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2021, 118, e2111863118. [Google Scholar] [CrossRef]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Canteli, M.; Iadecola, C. Alzheimer’s disease and vascular aging. J. Am. Coll. Cardiol. 2020, 75, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction-the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Huffman, K. The developing, aging neocortex: How genetics and epigenetics influence early developmental patterning and age-related change. Front. Genet. 2012, 3, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treder, M.S.; Chares, I.; Michelmann, S.; Martin-Buro, M.C.; Roux, F.; Carceller-Benito, F.; Ugalde-Canitrot, A.; Rollings, D.T.; Sawlani, V.; Chelvarajah, R.; et al. The hippocampus as the switchboard between perception and memory. Proc. Natl. Acad. Sci. USA 2021, 118, e2114171118. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hipocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef] [PubMed]
- Beckman, D.; Ott, S.; Donius-Cox, K.; Janssen, W.G.; Bliss-Moreau, E.; Rudebeck, P.H.; Baxter, M.G.; Morrison, J.H. Oligomeric Aß in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc. Natl. Acad. Sci. USA 2019, 116, 26239–26246. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Kang, S.S.; Wang, Z.; Liu, X.; Kuo, T.-C.; Korkmaz, F.; Padilla, A.; Miyashita, S.; Chan, P.; Zhang, Z.; et al. FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 2022, 603, 470–476. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aß plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.D.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, eabb8255. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Guo, J.L.; McBridge, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-ß plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Lee, M.-S.; Tsai, L.-H. Cdk5: One of the links between senile plaques and neurofibrollary tangles? J. Alzheimers Dis. 2003, 5, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Korte, N.; Nortley, R.; Attwell, D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropath. 2020, 140, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69. [Google Scholar] [CrossRef]
- Brown, G.C.; St George-Hyslop, P.H. Deciphering miocroglial diversity in Alzheimer’s disease. Science 2017, 356, 1123–1124. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellivol, M.; Brown, G.C. Microglia phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia- derived ASC specks cross-seed amyloid-ß in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.-Y.; Frost, G.F.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Park, J.; Griciuc, A.; Kim, E.; Choi, S.H.; Iwamoto, Y.; Kiss, M.G.; Christie, K.A.; Vinegoni, C.; Poller, W.C.; et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 2021, 595, 701–706. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katz-Marski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; Ezerskiy, L.A.; Morhaus, M.M.; Bannon, R.N.; Sauerbeck, A.D.; Shabsovich, M.; Jafar-nejad, P.; Rigo, F.; Miller, T.M. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2100356118. [Google Scholar] [CrossRef]
- Blennow, K. Phenotyping Alzheimer’s disease with blood tests. Science 2021, 373, 626–628. [Google Scholar] [CrossRef]
- Fagan, A.; Xiong, C.X.; Jasielec, M.S.; Bateman, R.J.; Goate, A.M.; Benzinger, T.L.S.; Ghetti, B.; Martins, R.N.; Masters, C.L.; Mayeux, R.; et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci. Transl. Med. 2014, 6, 226ra30. [Google Scholar] [CrossRef] [Green Version]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munij, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Broce, I.J.; Tan, C.H.; Fan, C.C.; Jansen, I.; Savage, J.E.; Witoelar, A.; Wen, N.; Hess, C.P.; Dillon, W.P.; Glastonbury, C.M.; et al. Dissecting the genetic relationship between cardiovascular risk factors and Alzheimer’s disease. Acta Neuropath. 2019, 137, 209–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural. Transm. 2002, 109, 813–836. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R., Jr. Cerebral amyloid angiopathy: Diagnosis, clinical implications, and management strategies in atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1173–1182. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.F.; Nicoli, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-ß pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zlokovic, B.V. Lymphatic waste disposal in the brain. Nature 2018, 560, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer’s disease-insights from amyloid-ß metabolism beyond the brain. Nat. Rev. 2017, 13, 612–623. [Google Scholar]
- Wiesmann, M.; Zerbi, V.; Jansen, D.; Lütjohann, D.; Veltin, A.; Heerschap, A.; Kiliaan, A.J. Hypertension, cerebrovascular impairment, and cognitive decline in aged AßPP/PS1 mice. Theranostics 2017, 7, 1277–1289. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral ß-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Aß-containing inoculates induce cerebral ß-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Guo, Q.; Inoue, T.; Polito, V.A.; Tabuchi, K.; Hammer, R.E.; Pautler, R.G.; Taffet, G.E.; Zheng, H. Vascular and parenchymal amyloid pathology in an Alzheimer’s disease knock-in mouse model: Interplay with cerebral blood flow. Mol. Neurodegener. 2014, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, G.; Adams, M.E.; Jaunmuktane, Z.; Lammie, G.A.; Turner, B.; Wani, M.; Sawhney, I.M.S.; Houlden, H.; Mead, S.; Brandner, S.; et al. Early onset cerebral amyloid angiopathy following childhood exposure to cadaveric dura. Ann. Neurol. 2019, 85, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Jiang, H.; Serrano, J.R.; Gonzales, E.R.; Wang, C.; Gratuze, M.; Hoyle, R.; Bien-Ly, N.; Silverman, A.P.; Sullivan, P.M.; et al. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med. 2021, 13, eabd7522. [Google Scholar] [CrossRef]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Maier, F.C.; Wehrl, H.F.; Schmid, A.M.; Mannheim, J.G.; Wiehr, S.; Lerdkrait, C.; Calaminus, C.; Stahlschmidt, A.; Ye, L.; Burnet, M.; et al. Longitudinal PET-MRI reveals ß-amyloid deposition and rCBF dynamics and connects vascular amyloidosis to quantitative loss of perfusion. Nat. Med. 2014, 20, 1485–1492. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Perez, J.M.; Evans, A.C. The Alzheimer’s Disease Neuroimaging Initiative (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Wolters, F.J.; Zonneveld, H.I.; Hofman, A.; van der Lugt, A.; Koudstaal, P.J.; Vernooij, M.W.; Ikram, M.A. Cerebral perfusion and the risk of dementia. Circulation 2017, 136, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Debbins, J.P.; Malek-Ahmadi, M.; Chen, K.; Pipe, J.G.; Maze, S.; Belden, C.; Maarouf, C.L.; Thiyyagura, P.; Mo, H.; et al. Cerebral blood flow in Alzheimer’s disease. Vasc. Health Risk Manag. 2012, 8, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid ß oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Preische, O.; Sohrabi, H.R.; Gräber, S.; Jucker, M.; Ringman, J.M.; Martins, R.N.; McDade, E.; Schofield, P.R.; Ghetti, B.; et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361, 991. [Google Scholar] [CrossRef] [Green Version]
- Asslani, I.; Habeck, C.; Scarmeas, N.; Borodovac, A.; Brown, T.R.; Stern, Y. Multivariate and univariate analysis of continuous arterial spin labeling perfusion MRI in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2008, 28, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Cruz Hernandez, J.C.; Bracko, O.; Kersbergen, C.J.; Muse, V.; Haft-Javaherian, B.M.; Park, L.; Vinarcsik, L.K.; Ivasyk, I.; Rivera, D.A. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat. Neurosci. 2019, 22, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activated processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [Green Version]
- Marshall, R.S.; Lazar, R.M.; Pile-Spellman, J.; Young, W.L.; Duong, D.H.; Joshi, S.; Ostapkovich, N. Recovery of brain function during induced cerebral hypoperfusion. Brain 2001, 124, 1208–1217. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Du, Y.; Wang, K.; Xu, G.; Luo, S.; He, G. Chronic cerebral hypoperfusion induces memory deficits and facilitates Aß gereration in C57BL/6J mice. Exp. Neurol. 2016, 283, 353–364. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Kruyer, A.; Fernandez-Nueda, I.; Marcos-Diaz, A.; Ceron, C.; Richards, A.T.; Jno-Charles, O.C.; Rodriguez, I.; Callejas, S.; Norris, E.; et al. Long-term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J. Am. Coll. Cardiol. 2019, 74, 1910–1923. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Chen, Z.-L.; Strickland, S.; Norris, E.H. Increased contact system activation in mild cognitive impairment patients with impaired short-term memory. J. Alzheimers Dis. 2020, 77, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Renne, T.; Strickland, S. The Alzheimer’s disease peptide ß-amyloid promotes thrombin generation through activation of coagulation factor XII. J. Thromb. Haemost. 2016, 14, 995–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamolodchikov, D.; Strickland, S. A possible new role for Aß in vascular and inflammatory dysfunction in Alzheimer’s disease. Thromb. Res. 2016, 141 (Suppl. S2), S59–S61. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Chen, Z.-L.; Conti, B.A.; Renne, T.; Strickland, S. Activation of the factor XII-driven contact system in Alzheimer’s disease patient and mouse model plasma. Proc. Natl. Acad. Sci. USA 2015, 112, 4068–4073. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K.; Chen, Z.-L.; Ghosh, D.; Strickland, S.; Norris, E.H. Increased plasma bradykinin level is associated with cognitive impairment in Alzheimer’s patiens. Neurobiol. Dis. 2020, 139, 1–8. [Google Scholar] [CrossRef]
- Chen, Z.-L.; Revenko, A.S.; Singh, P.; MacLeod, A.; Norris, E.H.; Strickland, S. Depletion of coagulation factor XII ameliorates brain pathology and cognitive impairment in Alzheimer’s disease mice. Blood 2017, 129, 2547–2556. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and ß-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide ß-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef] [Green Version]
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: Implications for Alzheimer’s disease. Front. Neurosci. 2020, 14, 762. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between ß-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes-Canteli, M.; Mattei, L.; Richards, A.T.; Norris, E.H.; Strickland, S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol. Aging 2015, 36, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultman, K.; Strickland, S.; Norris, E.H. The APOE e4/e4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cerebral. Blood Flow Metab. 2013, 33, 1251–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Z.; Yamashita, T.; Shi, X.; Feng, T.; Yu, H.; Hu, X.; Hu, X.; Bian, Y.; Sun, H.; Tadokoro, K.; et al. Accelerated accumulation of fibrinogen peptide chains with Aß deposition in Alzheimer’s disease (AD) mice and human brains. Brain Res. 2021, 1767, 147569. [Google Scholar] [CrossRef]
- Grammas, P.; Pezhman Ghatreh, S.; Lakshmi, T. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef]
- Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J.; Martinez, J.; Grammas, P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 2013, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; Rafalski, V.A.; Meyer-Franke, A.; Adams, R.A.; Poda, S.B.; Rios Coronado, P.E.; Ostergaard Pedersen, L.; Menon, V.; Baeten, K.M.; Silkorski, S.L.; et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 2018, 19, 1212–1223. [Google Scholar] [CrossRef]
- Silva, L.M.; Doyle, A.D.; Greenwell-Wild, T.; Dutzan, N.; Tran, C.L.; Abusleme, L.; Juang, L.J.; Leung, J.; Chun, E.M.; Lum, A.G.; et al. Fibrin is a critical regulator of neutrophil effector function at the oral mucosal barrier. Science 2021, 374, 1575. [Google Scholar] [CrossRef]
- Tang, M.-Y.; Gorin, F.A.; Lein, P.J. Review of evidence implicating the plasminogen activator system in blood-brain barrier dysfunction associated with Alzheimer’s disease. Ageing. Neur. Dis. 2022, 2, 2. [Google Scholar] [CrossRef]
- Cajamarca, S.A.; Norris, E.H.; van der Weerd, L.; Strickland, S.; Ahn, H.J. Cerebral amyloid angiopathy-linked ß-amyloid mutations promote cerebral fibrin deposits via increased binding affinity to fibrinogen. Proc. Natl. Acad. Sci. USA 2020, 117, 14482–14492. [Google Scholar] [CrossRef]
- van Oijen, M.; Witteman, J.C.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M.B. Fibrinogen is associated with increased risk of Alzheimer Disease and vascular dementia. Stroke 2005, 36, 2637–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, H.J.; Glickman, J.F.; Poon, K.L.; Zamolodchikov, D.; Jno-Charles, O.C.; Norris, E.H.; Strickland, S. A novel Aß-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J. Exp. Med. 2014, 211, 1049–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamaschini, L.; Rossi, E.; Storini, C.; Pizzimenti, S.; Distaso, M.; Perego, C.; De Luigi, A.; Vergani, C.; De Simoni, M.G. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and ß-amyloid accumulation in a mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 4181–4186. [Google Scholar] [CrossRef] [PubMed]
- Timmer, N.M.; van Dijk, L.; van der Zee, C.E.E.M.; Kiliaan, A.; de Waal, R.M.W.; Verbeek, M.M. Enoxaparin treatment administered at both early and late stages of amyloid ß deposition improves cognition of AOOswe/PS12dE9 mice with differential effects on brain Aß levels. Neurobiol. Dis. 2010, 40, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, M.N.; Braun, D.; Situ, A.; Moyano, A.L.; Kalinin, S.; Polak, P.; Givogri, M.; Feinstein, D.L. Differential effects on glial activation by a direct versus indirect thrombin inhibitor. J. Neuroimmunol. 2016, 297, 159–168. [Google Scholar] [CrossRef]
- Khalil, R.B. Direct thrombin inhibitor’s potential efficacy in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Dement. 2012, 27, 564–567. [Google Scholar]
- Whittier, J.R.; Korenyi, C.; Klein, D.F.; Foley, W. Prevention of degenerative disease: A controlled study of anticoagulant prophylaxis. J. Chronic Dis. 1961, 14, 203–212. [Google Scholar] [CrossRef]
- Ratner, J.; Rosenberg, G.; Kral, V.A.; Engelsmann, F. Anticoagulant therapy for senile dementia. J. Am. Geriatr. Soc. 1972, 20, 556–559. [Google Scholar] [CrossRef]
- Walsh, A.C.; Walsh, B.H.; Melaney, C. Senile-presenile dementia: Follow-up data on an effective psychotherapy-anticoagulant regimen. J. Am. Geriatr. Soc. 1978, 26, 467–470. [Google Scholar] [CrossRef]
- Barber, M.; Tait, C.; Scott, J.; Rumley, A.; Lowe, D.O.; Stott, D.J. Dementia in subjects with atrial fibrillation: Hemostatic function and the role of anticoagulation. J. Throm. Haemost 2004, 2, 1873–1878. [Google Scholar] [CrossRef]
- Friberg, L.; Rosenqvist, M. Less dementia with oral anticoagulation in atrial fibrillation. Eur. Heart J. 2018, 39, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongkhon, P.; Fanning, L.; Lau, W.C.Y.; Tse, G.; Lau, K.K.; Wei, L.; Kongkaew, C.; Wong, I.C.K. Oral anticoagulant and reduced risk of dementia in patients with atrial fibrillation: A population-based cohort study. Heart Rhythm 2020, 17, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, V.; May, H.T.; Bair, T.L.; Crandall, B.G.; Cutler, M.J.; Day, J.D.; Mallender, C.; Osborn, J.S.; Stevens, S.M.; Weiss, J.P.; et al. Long-term population-based cerebral ischemic event and cognitive outcomes of direct oral anticoagulants compared with warfarin among long-term anticoagulated patients for atrial fibrillation. Am. J. Cardiol. 2016, 118, 210–214. [Google Scholar] [CrossRef]
- Cadogan, S.L.; Powell, E.; Wing, K.; Wong, A.Y.; Smeeth, L.; Warren-Gash, C. Anticoagulant prescribing for atrial fibrillation and risk of incident dementia. Heart 2021, 107, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liu, W.; Li, B.; Li, D. Relationship of anticoagulant therapy with cognitive impairment among patients with atrial fibrillation. A meta-analysis and systemic review. J. Cardiovasc. Pharmocol. 2018, 71, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Jiang, C.; Su, C.; Tan, Y.; Wu, J. Anticoagulation in atrial fibrillation and cognitive decline. Medicine 2019, 98, e14499. [Google Scholar] [CrossRef] [PubMed]
- Mongkhon, P.; Naser, A.Y.; Fanning, L.; Tse, G.; Lau, W.C.Y.; Wong, I.C.K.; Kongkaew, C. Oral anticoagulants and risk of dementia: A systematic review and meta-analysis of observational studies and randomized controlled trials. Neurosci. Biobehav. Rev. 2019, 96, 1–9. [Google Scholar] [CrossRef]
- Ho, B.-L.; Hsieh, S.-W.; Chou, P.-S.; Yang, Y.-H. Effects of dabigatran on dementia pathogenesis and neuropsychological function: A review. J. Alzheimers Dis. 2022, 86, 1589–1601. [Google Scholar] [CrossRef]
- Bunch, T.J.; May, H.T.; Cutler, M.J.; Woller, S.C.; Jacobs, V.; Stevens, S.; Carlquist, J.; Knowlton, K.U.; Muhlestein, J.B.; Steinberg, B.A.; et al. Impact of anticoagulation therapy on the cognitive decline and dementia in patients with non-valvular atrial fibrillation (CAF) Trial. J. Am. Coll. Cardiol. 2022, 79, 9. [Google Scholar] [CrossRef]
- Rivard, L.; Khairy, P.; Talajic, M.; Tardif, J.-C.; Nattel, S.; Bherer, L.; Black, S.; Healey, J.; Lanthier, S.; Andrade, J.; et al. Blinded randomized trial of anticoagulation to prevent ischemic stroke and neurodegenerative impairment in AF (BRAIN-AF): Methods and design. Can. J. Cardiol. 2019, 35, 1069–1077. [Google Scholar] [CrossRef]
- Bates, S.M.; Weitz, J.I. The mechanism of action of thrombin inhibitors. J. Invasive Cardiol. 2000, 12, F27–F32. [Google Scholar]
- Monreal, M.; Costa, J.; Salva, P. Pharmacological properties of hirudin and its derivatives. Potential clinical advantages over heparin. Drugs Aging 1996, 8, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Q.; Zhou, Y.-P.; Yang, H. Donepezil combined with natural hirudin improves the clinical symptoms of patients with mild-to-moderate Alzheimer’s disease: A 20-week open-label pilot study. Int. J. Med. Sci. 2012, 9, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zirlik, A.; Bode, C. Vitamin K antagonists: Relative strengths and weaknesses vs. direct oral anticoagulants for stroke prevention in patients with atrial fibrillation. J. Thromb. Thrombolysis 2017, 43, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferland, G. Vitamin K and the nervous system: An overview of its actions. Adv. Nut. 2012, 3, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Van Ryn, J.; Goss, A.; Hauel, N.; Wienen, W.; Priepke, H.; Nar, H.; Clemens, A. The discovery of Dabigatran etexilate. Front. Pharmacol. 2013, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Lip, G.Y.H.; Keshishian, A.; Li, X.; Hamilton, M.; Masseria, C.; Gupta, K.; Luo, X.; Mardekian, J.; Friend, K.; Nadkarni, A.; et al. Effectiveness and safety of oral anticoagulants among nonvalvular atrial fibrillation patients. The ARISTOPHANES Study. Stroke 2018, 49, 2933–2944. [Google Scholar] [CrossRef] [Green Version]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- Fanning, L.; Lau, W.C.Y.; Mongkhon, P.; Man, K.K.C.; Bell, J.S.; Ilomäki, J.; Darzins, P.; Lau, K.K.; Wei, L.; Wong, I.C.K. Safety and effectiveness of direct oral anticoagulants vs warfarin in people with atrial fibrillation and dementia. J. Am. Med. Dir. Assoc. 2020, 21, 1058–1064. [Google Scholar] [CrossRef]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.V.; Lopes, M.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A.; et al. Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.J.; Reichman, M.E.; Wernecke, M.; Zhang, R.; Southworth, M.R.; Levenson, M.; Sheu, T.-C.; Mott, K.; Goulding, M.R.; Houstoun, M.; et al. Cardiovascular, bleeding, and mortality risks in elderly medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation 2015, 131, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.J.; Reichman, M.E.; Wernecke, M.; Hsueh, Y.-H.; Izem, R.; Southworth, M.R.; Wei, Y.; Liao, J.; Goulding, M.R.; Mott, K.; et al. Stroke, bleeding, and mortality risks in elderly medicare beneficiaries treated with dabigatran or rivaroxaban for nonvalvular atrial fibrillation. JAMA Int. Med. 2016, 176, 1662–1671. [Google Scholar]
- Marinescu, M.; Sun, L.; Fatar, M.; Neubauer, A.; Schad, L.; van Ryn, J.; Lehmann, L.; Veltkamp, R. Cerebral microbleed in murine amyloid angiopathy. Natural course and anticoagulant effects. Stroke 2017, 48, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Michael, N.; Grigoryan, M.M.; Kilday, K.; Sumbria, R.K.; Vasilevko, V.; van Ryn, J.; Cribbs, D.H.; Paganini-Hill, A.; Fisher, M.J. Effects of dabigatran in mouse models of aging and cerebral amyloid angiopathy. Front. Neurol. 2019, 10, 966. [Google Scholar] [CrossRef]
- Pollack, C.V., Jr.; Paul, M.D.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Hulsman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for dabigatran reversal. N. Engl. J. Med. 2015, 373, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.J.; Truman, J.M., Jr.; Eikelboom, J.W.; Gibson, C.M.; Curnutte, J.T.; Gold, A.; Bronson, M.D.; Lu, G.; Conley, P.B.; Verhamme, P.; et al. for the ANNEXA-4 investigators Andexanet alfa for acute major bleeding associated with factor Xa inhibitors. N. Engl. J. Med. 2016, 375, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Bogatkevich, G.S.; Ludwicka-Bradley, A.; Nietert, P.J.; Akter, T.; van Ryn, J.; Silver, R.M. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum. 2011, 63, 1416–1425. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Novel Therapeutic Target for Alzheimer’s Disease in Men and Women 50-85 Years of Age. Available online: https://clinicaltrials.gov/ct2/show/NCT03752294 (accessed on 8 June 2022).
- Greenberg, S.M.; Charidimou, A. Diagnosis of cerebral amyloid angiopathy. Evolution of the Boston criteria. Stroke 2018, 49, 491–497. [Google Scholar] [CrossRef]
- Adrover, J.M.; Pellico, J.; Fernandez-Barahona, I.; Martin-Salamanca, S.; Ruiz-Cabello, J.; Hidalgo, A.; Herranz, F. Thrombo-tag, an in vivo formed nanotracer for the detection of thrombi in mice by fast pre-targeted molecular imaging. Nanoscale 2020, 12, 22978–22987. [Google Scholar] [CrossRef]
- Dai, W.; Lopez, O.L.; Carmichael, O.T.; Becker, J.T.; Kuller, L.H.; Gach, H.M. Mild cognitive impairment and Alzheimer’s disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 2009, 250, 856–866. [Google Scholar] [CrossRef]
- Dolgin, E. A tough spot. Nature 2018, 559, S10–S12. [Google Scholar] [CrossRef] [PubMed]
- Mankhong, S.; Kim, S.; Lee, S.; Kwak, H.-B.; Park, D.-H.; Joa, K.-L.; Kang, J.-H. Development of Alzheimer’s disease biomarkers: From CSF- to blood-based biomarkers. Biomedicines 2022, 10, 850. [Google Scholar] [CrossRef] [PubMed]
- Eninger, T.; Müller, S.A.; Bacioglu, M.; Schweighauser, M.; Lambert, M.; Maia, L.F.; Neher, J.J.; Hornfeck, S.M.; Obermüller, U.; Kleinberger, G.; et al. Signatures of glial activity can be detected in the CSF proteome. Proc. Natl. Acad. Sci. USA 2022, 119, e2119804119. [Google Scholar] [CrossRef] [PubMed]
- Beyer, L.; Stocker, H.; Rujescu, D.; Holleczek, B.; SStockmann, J.; Nabers, A.; Brenner, H.; Gerwert, K. Amyloid-beta misfolding and GFAP predict risk of clinical Alzheimer’s disease diagnosis within 17 years. Alzheimer’s Dement. 2022, 1–9. [Google Scholar] [CrossRef]
- Islam, M.R.; Kaurani, L.; Berulava, T.; Heilbronner, U.; Budde, M.; Centeno, T.P.; Elerdashvili, V.; Zafieriou, M.-P.; Benito, E.; Sertel, S.M.; et al. A microRNA signature that correlates with cognition and is a target against cognitive decline. EMBO Mol. Med. 2021, 13, e13659. [Google Scholar] [CrossRef]
- Laske, C.; Müller, S.; Preische, O.; Ruschil, V.; Munk, M.H.J.; Honold, I.; Peter, S.; Schoppmeier, U.; Willmann, M. Signature of Alzheimer’s disease in intestinal microbiome: Results from the AlzBiom study. Front. Neurosci. 2022, 16, 792996. [Google Scholar] [CrossRef]
- Purgatorio, R.; Gambacorta, N.; de Candida, M.; Catto, M.; Rullo, M.; Pisani, L.; Nicolotti, O.; Altomare, C.D. First-in-class isonipecotamide-based thrombin and cholinesterase dual inhibitors with potential for Alzheimer disease. Molecules 2021, 26, 5208. [Google Scholar] [CrossRef]
- Elabi, O.; Gaceb, A.; Carlsson, R.; Padel, T.; Soylu-Kucharz, R.; Cortijo, I.; Li, W.; Li, J.-Y.; Paul, G. Human α-synuclein overexpression in a mouse model of Parkinson’s disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Nat. Res. 2021, 11, 1120. [Google Scholar] [CrossRef]
- Garcia, F.J.; Sun, N.; Lee, H.; Godlewski, B.; Mathys, H.; Galani, K.; Zhou, B.; Jiang, Y.; Ng, A.P.; Mantero, J.; et al. Single-cell dissection of the human brain vasculature. Nature 2022, 603, 893–899. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grossmann, K. Direct Oral Anticoagulants (DOACs) for Therapeutic Targeting of Thrombin, a Key Mediator of Cerebrovascular and Neuronal Dysfunction in Alzheimer’s Disease. Biomedicines 2022, 10, 1890. https://doi.org/10.3390/biomedicines10081890
Grossmann K. Direct Oral Anticoagulants (DOACs) for Therapeutic Targeting of Thrombin, a Key Mediator of Cerebrovascular and Neuronal Dysfunction in Alzheimer’s Disease. Biomedicines. 2022; 10(8):1890. https://doi.org/10.3390/biomedicines10081890
Chicago/Turabian StyleGrossmann, Klaus. 2022. "Direct Oral Anticoagulants (DOACs) for Therapeutic Targeting of Thrombin, a Key Mediator of Cerebrovascular and Neuronal Dysfunction in Alzheimer’s Disease" Biomedicines 10, no. 8: 1890. https://doi.org/10.3390/biomedicines10081890
APA StyleGrossmann, K. (2022). Direct Oral Anticoagulants (DOACs) for Therapeutic Targeting of Thrombin, a Key Mediator of Cerebrovascular and Neuronal Dysfunction in Alzheimer’s Disease. Biomedicines, 10(8), 1890. https://doi.org/10.3390/biomedicines10081890