
The Multifaceted Roles of Autophagy in Infectious, Obstructive, and Malignant Airway Diseases

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Autophagy Overview
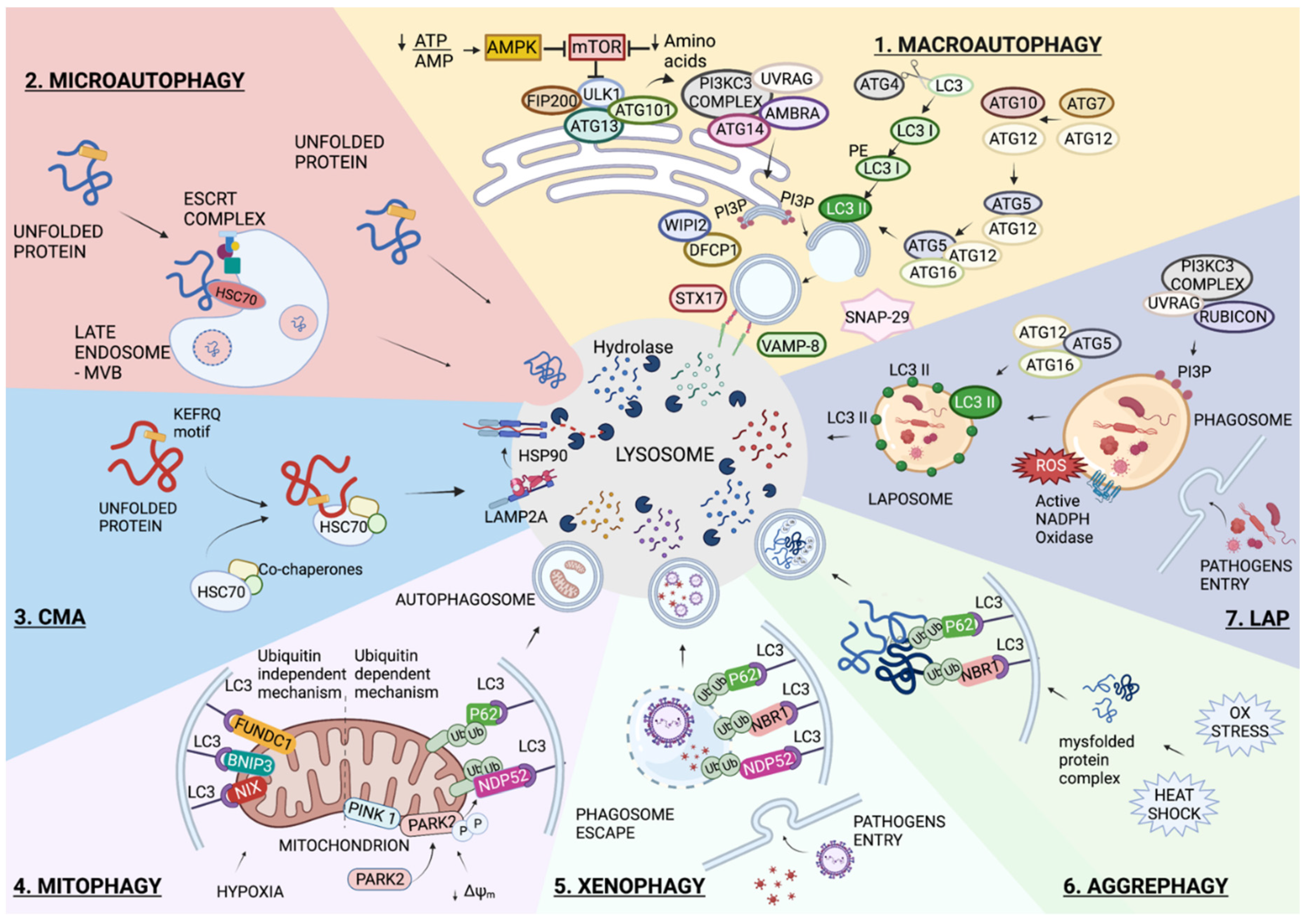
1.1.1. Macroautophagy
1.1.2. Microautophagy
1.1.3. Chaperone-Mediated Autophagy (CMA)
1.1.4. Selective Autophagy
Mitophagy
- (i).
- Mitochondrial depolarization causes the accumulation of mitochondrial PTEN Induced Kinase 1 (PINK1) on the Outer Mitochondrial Membrane (OMM) [28,29]. The activation of PINK1 through its auto-phosphorylation starts mitophagy in two parallel processes: (a) the phosphorylation of ubiquitin at serine 65 and (b) the phosphorylation of Parkin RBR E3 Ubiquitin Protein Ligase (PARK2) [30]. The interaction of phosphorylated PARK2 with phospho-ubiquitin on mitochondria results in PARK2 activation, which is responsible for the ubiquitination of OMM proteins [31,32]. This process permits the recruitment of autophagic receptors to the OMM, which links ubiquitylated proteins with autophagosomes via their ubiquitin-binding domains and LC3-Interacting Region (LIR) motifs, respectively [33].
- (ii).
- The LC3 receptors on the mitochondrial membrane directly bind to LC3 and play an important role in the recruitment of damaged mitochondria to the autophagosomes. These receptors, including NIX, BNIP3, and FUNDC1, induce mitophagy under hypoxia conditions [34,35,36]. Bcl-2 like 13 (Bcl-2L13) and FKBP Prolyl Isomerase 8 (FKBP8) are located on the OMM and mediate mitophagy by interacting with LC3, via the LIR motif [37] and via LC3A [38], respectively.
Xenophagy
Aggrephagy
1.1.5. LC3-Associated Phagocytosis (LAP)
2. Autophagy in Lung Infections
2.1. Autophagy in SARS-CoV-2 Infection
2.1.1. Exploiting Autophagy by SARS-CoV-2
2.1.2. Targeting Autophagy in SARS-CoV-2 Infection
2.2. Autophagy in Respiratory Syncytial Virus (RSV) Infection
2.2.1. Autophagy Activation in Intracellular Elimination of RSV
2.2.2. Exploitation of Autophagy by RSV
2.2.3. Targeting Host Autophagy to Counteract RSV Infection
2.3. Autophagy in Mycobacterium Tuberculosis (Mtb) Infection
2.3.1. Autophagy Activation in the Intracellular Elimination of Mtb
2.3.2. Exploiting Autophagy by Mtb
2.3.3. Targeting Host Autophagy to Counteract Mtb Infection
3. Autophagy in Lung Diseases
3.1. Autophagy in Cystic Fibrosis (CF)
3.1.1. Dysregulation of Autophagy in CF
3.1.2. Targeting Autophagy in CF
3.2. Autophagy in Chronic Obstructive Pulmonary Disease (COPD)
3.2.1. Dysregulation of Autophagy in COPD
3.2.2. Targeting Autophagy in COPD
3.3. Autophagy in Malignant Mesothelioma (MM)
3.3.1. Dysregulation of Autophagy in MM
3.3.2. Targeting Autophagy in MM
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AICAR | AMP-mimetic 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside |
| AM | Alveolar Macrophages |
| AMBRA1 | Activating Molecule in BECN1-Regulated Autophagy protein 1 |
| AMPK | 5′ AMP-Activated Protein Kinase |
| ATGs | Autophagic-Related Genes |
| BAP1 | BRCA1 Associated Protein 1 |
| BECN1 | Beclin 1 |
| BNIP3 | Bcl-2 Interacting Protein 3 |
| BNIP3L/NIX | Bcl-2 Interacting Protein 3 Like |
| CF | Cystic Fibrosis |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| cGAS | cyclic GMP-AMP synthase |
| CMA | Chaperone-Mediated Autophagy |
| COPD | Chronic Obstructive Pulmonary Disease |
| CS | Cigarette smoke |
| CSE | Cigarette smoke extract |
| DCs | Dendritic cells |
| DFCP1 | Zinc Finger FYVE-Type Containing 1 |
| DRP1 | Dynamin 1 Like |
| Egr-1 | Early growth response-1 |
| Eis | Enhanced intracellular survival |
| Ephx2 | Soluble epoxide hydrolase |
| ER | Endoplasmic Reticulum |
| ESCRT | Endosomal Sorting Complexes Required for Transport |
| ESX-1 | ESAT-6 Secretion System-1 |
| ESX-1 | ESAT-6 Secretion System-1 |
| FAM134B | Family With Sequence Similarity 134 Member B |
| FIP200 | RIB-inducible coiled-coil protein 1 |
| FUNDC1 | FUN14 Domain Containing 1 |
| GABARAP | Gamma-Aminobutyric Acid Receptor-Associated Protein |
| GSNO | S-Nitrosoglutathione |
| HDAC6 | Histone Deacetylase 6 |
| HDT | Host directed therapy |
| HMGB1 | High Mobility Group Box 1 |
| HSC70 | Heat Shock Cognate 71 kDa Protein |
| IFN | Interferon |
| IL | Interleukin |
| LAMP2A | Lysosome-Associated Membrane Protein type 2A |
| LAP | LC3-associated phagocytosis |
| LC3 | Microtubule-Associated Protein 1A/B Light-Chain 3 |
| LIR | LC3-Interacting Region |
| LTB4 | Leukotriene B4 |
| MAVS | Mitochondrial Antiviral-Signaling Protein |
| MCC | Mucociliary clearance |
| MCU | Mitochondrial Calcium Uniporter |
| Mtb | Mycobacterium tuberculosis |
| mTOR | Mechanistic target of rapamycin |
| NBR1 | Neighbor of BRCA1 Gene 1 Protein |
| NCOA4 | Nuclear Receptor Coactivator 4 |
| NDP52 | Calcium Binding And Coiled-Coil Domain 2 |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| NO | Nitric Oxide |
| NOX2 | NADPH oxidase-2 |
| Nrf2 | NFE2 Like BZIP Transcription Factor 2 |
| OMM | Outer Mitochondrial Membrane |
| OPTN | Optineurin |
| ORF | Open Reading Frame |
| P2X7 | Purinergic Receptor P2X 7 |
| p62/SQSTM1 | Sequestome |
| PAFR | Platelet-Activating Factor Receptor |
| PAMPs_viral | Pathogen-Associated Molecular Patterns |
| PARK2 | Parkin RBR E3 Ubiquitin Protein Ligase |
| PE | Phosphatidylethanolamine |
| PI3KC3 | Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3 |
| PI3P | Phosphatidylinositol 3-Phosphate |
| PINK1 | PTEN Induced Kinase 1 |
| PM | Particulate Matter |
| PTPIP51 | Protein Tyrosine Phosphatase Interacting Protein 51 |
| QF | Qingfei |
| RAB7 | Ras-Associated Protein 7 |
| RIP3 | Receptor Interacting Serine/Threonine Kinase 3 |
| ROS | Reactive Species of Oxygen |
| RSV | Respiratory Syncytial Virus |
| Rubicon | RUN And Cysteine Rich Domain Containing Beclin 1 Interacting Protein |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus–2 |
| SIRT | Sirtuin |
| SKP2 | S-Phase Kinase Associated Protein 2 |
| SNAP-29 | Synaptosome Associated Protein 29 |
| SNARE | Soluble N-ethylmaleimide Sensitive Fusion Protein (NSF) Attachment Receptor |
| STING | Stimulator of Interferon Response CGAMP Interactor 1 |
| STX17 | Syntaxin 17 |
| TAX1BP1 | Tax1 Binding Protein 1 |
| TB | Tuberculosis |
| TFEB | Transcription Factor EB |
| TG2 | Transglutaminase 2 |
| ULK1 | Unc-51-Like Kinase 1 |
| UPR | Unfolding protein response |
| UVRAG | UV radiation resistance-associated gene protein |
| VAMP-8 | Vesicle Associated Membrane Protein 8 |
| VAPB | Vesicle-Associated Membrane Protein-Associated Protein B |
| VPS | Vacuolar Protein Sorting |
| WIPI2 | WD Repeat Domain, Phosphoinositide Interacting 2 |
References
- Reggio, A.; Buonomo, V.; Grumati, P. Eating the Unknown: Xenophagy and ER-Phagy Are Cytoprotective Defenses against Pathogens. Exp. Cell Res. 2020, 396, 112276. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-Independent Alternative Macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Morshed, S.; Sharmin, T.; Ushimaru, T. TORC1 Regulates ESCRT-0 Complex Formation on the Vacuolar Membrane and Microautophagy Induction in Yeast. Biochem. Biophys. Res. Commun. 2020, 522, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the Selective Blockage of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.X.; Sun, P.P.; Gu, Y.H.; Rao, X.M.; Zhang, L.Y.; Ou-Yang, Y. Autophagy and Pulmonary Disease. Ther. Adv. Respir. Dis. 2019, 13, S40–S47. [Google Scholar] [CrossRef]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 Kinase Domain Dynamics Regulate the Autophagic PI 3-Kinase Complex. Mol. Cell 2017, 67, 528–534.e3. [Google Scholar] [CrossRef]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and Tumour Suppressor Activity of a Novel Beclin1-Binding Protein UVRAG. Nat. Cell Biol. 2006, 8, 688–698. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A Comprehensive Glossary of Autophagy-Related Molecules and Processes (2nd Edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 Links Autophagy to Cell Proliferation and Tumorigenesis by Promoting C-Myc Dephosphorylation and Degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.J.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG Conjugation Systems Are Important for Degradation of the Inner Autophagosomal Membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, X.; Ji, C.; Zhang, W.; Song, J.; Li, J.; Wang, J. Key Regulators of Autophagosome Closure. Cells 2021, 10, 2814. [Google Scholar] [CrossRef]
- Shen, Q.; Shi, Y.; Liu, J.; Su, H.; Huang, J.; Zhang, Y.; Peng, C.; Zhou, T.; Sun, Q.; Wan, W.; et al. Acetylation of STX17 (Syntaxin 17) Controls Autophagosome Maturation. Autophagy 2021, 17, 1157–1169. [Google Scholar] [CrossRef]
- Oku, M.; Maeda, Y.; Kagohashi, Y.; Kondo, T.; Yamada, M.; Fujimoto, T.; Sakai, Y. Evidence for ESC RT- and Clathrin-Dependent Microautophagy. J. Cell Biol. 2017, 216, 3263–3274. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Rapamycin Activates Mammalian Microautophagy. J. Pharmacol. Sci. 2019, 140, 201–204. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Hu, H.; Kshitiz Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-Mediated Autophagy Targets Hypoxia-Inducible Factor-1β(HIF-1β) for Lysosomal Degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A Role for a 70-Kilodaton Heat Shock Protein in Lysosomal Degradation of Intracellular Proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Dice, J.F. Peptide Sequences That Target Proteins for Enhanced Degradation during Serum Withdrawal. J. Biol. Chem. 1988, 263, 6797–6805. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, X.W.; Liu, Z.H.; Sun, Y.M.; Tang, Y.X.; Zhou, D.H. Chaperone-Mediated Autophagy Substrate Proteins in Cancer. Oncotarget 2017, 8, 51970–51985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular Definitions of Autophagy and Related Processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Vo, M.; Choi, Y. Herpesvirus Regulation of Selective Autophagy. Viruses 2021, 13, 820. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Variants of Mitochondrial Autophagy: Types 1 and 2 Mitophagy and Micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Ordureau, A.; Paulo, J.A.; Zhang, W.; Ahfeldt, T.; Zhang, J.; Cohn, E.F.; Hou, Z.; Heo, J.M.; Rubin, L.L.; Sidhu, S.S.; et al. Dynamics of PARKIN-Dependent Mitochondrial Ubiquitylation in Induced Neurons and Model Systems Revealed by Digital Snapshot Proteomics. Mol. Cell 2018, 70, 211–227.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauer, T.; Simicek, M.; Schubert, A.; Komander, D. Mechanism of Phospho-Ubiquitin-Induced PARKIN Activation. Nature 2015, 524, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade Harper, J.; Ordureau, A.; Heo, J.M. Building and Decoding Ubiquitin Hains for Mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-Mediated Mitophagy Protects against Ischemic Brain Injury Independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Shirihai, O.S.; Song, M.; Dorn, G.W. How Mitochondrial Dynamism Orchestrates Mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef] [Green Version]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.O.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [Green Version]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 Recruits LC3A to Mediate Parkin-independent Mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium Tuberculosis Survival in Infected Macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.H.; Song, H.K. A Structural View of Xenophagy, a Battle between Host and Microbes. Mol. Cells 2018, 41, 27–34. [Google Scholar] [CrossRef]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Y.; Wang, X.; Xing, R.; Liu, K.; Gan, Q.; Tang, C.; Gao, Z.; Jian, Y.; Luo, S.; et al. The BEACH-Containing Protein WDR81 Coordinates P62 and LC3C to Promote Aggrephagy. J. Cell Biol. 2017, 216, 1301–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Martinez, J. LAP It up, Fuzz Ball: A Short History of LC3-Associated Phagocytosis. Curr. Opin. Immunol. 2018, 55, 54–61. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular Characterization of LC3-Associated Phagocytosis Reveals Distinct Roles for Rubicon, NOX2 and Autophagy Proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, S.; Philips, J.A. LC3-Associated Phagocytosis: Host Defense and Microbial Response. Curr. Opin. Immunol. 2019, 60, 81–90. [Google Scholar] [CrossRef]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-Associated Protein 1 Light Chain 3 Alpha (LC3)-Associated Phagocytosis Is Required for the Efficient Clearance of Dead Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD 40 Domain of ATG 16L1 Is Required for Its Non-canonical Role in Lipidation of LC 3 at Single Membranes. EMBO J. 2018, 37, e97840. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Yang, C.S.; Lee, J.S.; Rodgers, M.; Min, C.K.; Lee, J.Y.; Kim, H.J.; Lee, K.H.; Kim, C.J.; Oh, B.; Zandi, E.; et al. Autophagy Protein Rubicon Mediates Phagocytic NADPH Oxidase Activation in Response to Microbial Infection or TLR Stimulation. Cell Host Microbe 2012, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Tosta, E. The Adaptation of SARS-CoV-2 to Humans. Mem. Inst. Oswaldo Cruz 2021, 116, e210127. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, X.; Zhu, Y.; Wang, W.; Wang, Y.; Hu, G.; Liu, C.; Li, J.; Ren, S.; Xiao, M.Z.X.; et al. ORF3a-Mediated Incomplete Autophagy Facilitates Severe Acute Respiratory Syndrome Coronavirus-2 Replication. Front. Cell Dev. Biol. 2021, 9, 716208. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Bozzo, C.P.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 Suppresses the Antiviral Innate Immune Response by Degrading MAVS through Mitophagy. Cell. Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Li, N.; et al. Inhibition of Autophagy Suppresses SARS-CoV-2 Replication and Ameliorates Pneumonia in HACE2 Transgenic Mice and Xenografted Human Lung Tissues. J. Virol. 2021, 95, 24. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 Protein ORF3a Inhibits Fusion of Autophagosomes with Lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardão, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The Mitochondrial Permeability Transition Pore: An Evolving Concept Critical for Cell Life and Death. Biol. Rev. 2021, 96, 2489–2521. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2022, 12, 780768. [Google Scholar] [CrossRef]
- Sun, X.; Yu, J.; Wong, S.H.; Chan, M.T.V.; Zhang, L.; Wu, W.K.K. SARS-CoV-2 Targets the Lysosome to Mediate Airway Inflammatory Cell Death. Autophagy 2022. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, C.L.; Venkatachalam, M.A.; Dong, Z. Autophagy Inhibition by Chloroquine and Hydroxychloroquine Could Adversely Affect Acute Kidney Injury and Other Organ Injury in Critically Ill Patients with COVID-19. Kidney Int. 2020, 98, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The Effect of Fluoxetine on Astrocyte Autophagy Flux and Injured Mitochondria Clearance in a Mouse Model of Depression. Cell Death Dis. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zschocke, J.; Zimmermann, N.; Berning, B.; Ganal, V.; Holsboer, F.; Rein, T. Antidepressant Drugs Diversely Affect Autophagy Pathways in Astrocytes and Neurons-Dissociation from Cholesterol Homeostasis. Neuropsychopharmacology 2011, 36, 1754–1768. [Google Scholar] [CrossRef]
- Hoertel, N.; Sánchez-Rico, M.; Vernet, R.; Beeker, N.; Jannot, A.S.; Neuraz, A.; Salamanca, E.; Paris, N.; Daniel, C.; Gramfort, A.; et al. Association between Antidepressant Use and Reduced Risk of Intubation or Death in Hospitalized Patients with COVID-19: Results from an Observational Study. Mol. Psychiatry 2021, 26, 5199–5212. [Google Scholar] [CrossRef]
- Rosen, D.A.; Seki, S.M.; Fernández-Castañeda, A.; Beiter, R.M.; Eccles, J.D.; Woodfolk, J.A.; Gaultier, A. Modulation of the Sigma-1 Receptor-IRE1 Pathway Is Beneficial in Preclinical Models of Inflammation and Sepsis. Sci. Transl. Med. 2019, 11, eaau5266. [Google Scholar] [CrossRef]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-Mediated Dysregulation of Metabolism and Autophagy Uncovers Host-Targeting Antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Dou, Q.; Chen, H.N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, M.L.; Li, Z.; Gao, D.M.; Cai, Y.; Chang, J.; Wang, S.P. Interferon-Alpha-2b Induces Autophagy in Hepatocellular Carcinoma Cells through Beclin1 Pathway. Cancer Biol. Med. 2014, 11, 64–68. [Google Scholar] [CrossRef]
- Hsu, H.C.; Chen, Y.H.; Lin, T.S.; Shen, C.Y.; Hsieh, S.C. Systemic Lupus Erythematosus Is Associated with Impaired Autophagic Degradation via Interleukin-6 in Macrophages. Biochim. Biophys. Acta-Mol. Basis Dis. 2021, 1867, 166027. [Google Scholar] [CrossRef]
- Collins, P.L.; Hill, M.G.; Cristina, J.; Grosfeld, H. Transcription Elongation Factor of Respiratory Syncytial Virus, a Nonsegmented Negative-Strand RNA Virus. Proc. Natl. Acad. Sci. USA 1996, 93, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory Syncytial Virus—A Comprehensive Review. Clin. Rev. Allergy Immunol. 2013, 45, 331–379. [Google Scholar] [CrossRef]
- Battles, M.B.; McLellan, J.S. Respiratory Syncytial Virus Entry and How to Block It. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.F.; Ikizler, M.R.; Gonzales, R.A.; Carroll, K.N.; Johnson, J.E.; Werkhaven, J.A. Growth of Respiratory Syncytial Virus in Primary Epithelial Cells from the Human Respiratory Tract. J. Virol. 2005, 79, 8651–8654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.A.; Colin, A.A. Infantile Respiratory Syncytial Virus and Human Rhinovirus Infections: Respective Role in Inception and Persistence of Wheezing. Eur. Respir. J. 2015, 45, 774–789. [Google Scholar] [CrossRef] [Green Version]
- Owczarczyk, A.B.; Schaller, M.A.; Reed, M.; Rasky, A.J.; Lombard, D.B.; Lukacs, N.W. Sirtuin 1 Regulates Dendritic Cell Activation and Autophagy during Respiratory Syncytial Virus–Induced Immune Responses. J. Immunol. 2015, 195, 1637–1646. [Google Scholar] [CrossRef]
- Morris, S.; Swanson, M.S.; Lieberman, A.; Reed, M.; Yue, Z.; Lindell, D.M.; Lukacs, N.W. Autophagy-Mediated Dendritic Cell Activation Is Essential for Innate Cytokine Production and APC Function with Respiratory Syncytial Virus Responses. J. Immunol. 2011, 187, 3953–3961. [Google Scholar] [CrossRef] [Green Version]
- Reed, M.; Morris, S.H.; Jang, S.; Mukherjee, S.; Yue, Z.; Lukacs, N.W. Autophagy-Inducing Protein Beclin-1 in Dendritic Cells Regulates CD4 T Cell Responses and Disease Severity during Respiratory Syncytial Virus Infection. J. Immunol. 2013, 191, 2526–2537. [Google Scholar] [CrossRef] [Green Version]
- Liesman, R.M.; Buchholz, U.J.; Luongo, C.L.; Yang, L.; Proia, A.D.; DeVincenzo, J.P.; Collins, P.L.; Pickles, R.J. RSV-Encoded NS2 Promotes Epithelial Cell Shedding and Distal Airway Obstruction. J. Clin. Investig. 2014, 124, 2219–2233. [Google Scholar] [CrossRef] [Green Version]
- Cortjens, B.; De Boer, O.J.; De Jong, R.; Antonis, A.F.G.; Sabogal Piñeros, Y.S.; Lutter, R.; Van Woensel, J.B.M.; Bem, R.A. Neutrophil Extracellular Traps Cause Airway Obstruction during Respiratory Syncytial Virus Disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef]
- Mukherjee, S.; Lindell, D.M.; Berlin, A.A.; Morris, S.B.; Shanley, T.P.; Hershenson, M.B.; Lukacs, N.W. IL-17Induced Pulmonary Pathogenesis during Respiratory Viral Infection and Exacerbation of Allergic Disease. Am. J. Pathol. 2011, 179, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Stoppelenburg, A.J.; Salimi, V.; Hennus, M.; Plantinga, M.; Huis in ’t Veld, R.; Walk, J.; Meerding, J.; Coenjaerts, F.; Bont, L.; Boes, M. Local IL-17A Potentiates Early Neutrophil Recruitment to the Respiratory Tract during Severe RSV Infection. PLoS ONE 2013, 8, e78461. [Google Scholar] [CrossRef] [PubMed]
- Ryzhakov, G.; Lai, C.C.; Blazek, K.; To, K.; Hussell, T.; Udalova, I. IL-17 Boosts Proinflammatory Outcome of Antiviral Response in Human Cells. J. Immunol. 2011, 187, 5357–5362. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, S.M.; Shil, N.K.; Bose, S. Autophagy, TGF-β, and SMAD-2/3 Signaling Regulates Interferon-β Response in Respiratory Syncytial Virus Infected Macrophages. Front. Cell. Infect. Microbiol. 2016, 6, 174. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, J.; Zeng, R.; Yang, J.; Liu, J.; Zhang, Z.; Song, X.; Yao, Z.; Ma, C.; Li, W.; et al. Respiratory Syncytial Virus Replication Is Promoted by Autophagy-Mediated Inhibition of Apoptosis. J. Virol. 2018, 92, e02193-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, H.; Levitz, R.; Huang, R.; Kahn, J.S. mTOR kinase is a therapeutic target for respiratory syncytial virus and coronaviruses. Sci. Rep. 2021, 11, 24442. [Google Scholar] [CrossRef]
- Lo, M.S.; Brazas, R.M.; Holtzman, M.J. Respiratory Syncytial Virus Nonstructural Proteins NS1 and NS2 Mediate Inhibition of Stat2 Expression and Alpha/Beta Interferon Responsiveness. J. Virol. 2005, 79, 9315–9319. [Google Scholar] [CrossRef] [Green Version]
- Sedeyn, K.; Schepens, B.; Saelens, X. Respiratory syncytial virus nonstructural proteins 1 and 2: Exceptional disrupters of innate immune responses. PLoS Pathog. 2019, 15, e1007984. [Google Scholar] [CrossRef]
- Han, B.; Wang, Y.; Zheng, M. Inhibition of autophagy promotes human RSV NS1-induced inflammation and apoptosis in vitro. Exp. Ther. Med. 2021, 22, 1054. [Google Scholar] [CrossRef]
- Chiok, K.; Pokharel, S.M.; Mohanty, I.; Miller, L.G.; Gao, S.J.; Haas, A.L.; Tran, K.C.; Teng, M.N.; Bose, S. Human Respiratory Syncytial Virus NS2 Protein Induces Autophagy by Modulating Beclin1 Protein Stabilization and ISGylation. MBio 2022, 13, e03528-21. [Google Scholar] [CrossRef]
- Chakraborty, S.; Castranova, V.; Perez, M.K.; Piedimonte, G. Nanoparticles increase human bronchial epithelial cell susceptibility to respiratory syncytial virus infection via nerve growth factor-induced autophagy. Physiol. Rep. 2017, 5, e13344. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Croix, C.S.; Good, M.; Chen, J.; Zhao, J.; Hu, S.; Ross, M.; Myerburg, M.M.; Pilewski, J.M.; Williams, J.; et al. Interleukin-22 Inhibits Respiratory Syncytial Virus Production by Blocking Virus-Mediated Subversion of Cellular Autophagy. iScience 2020, 23, 101256. [Google Scholar] [CrossRef] [PubMed]
- Bueno, S.M.; González, P.A.; Pacheco, R.; Leiva, E.D.; Cautivo, K.M.; Tobar, H.E.; Mora, J.E.; Prado, C.E.; Zúñiga, J.P.; Jiménez, J.; et al. Host Immunity during RSV Pathogenesis. Int. Immunopharmacol. 2008, 8, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Braciale, T.J. Respiratory Syncytial Virus Infection Suppresses Lung CD8+ T-Cell Effector Activity and Peripheral CD8+ T-Cell Memory in the Respiratory Tract. Nat. Med. 2002, 8, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, A.; Patton, K.; Gasser, R.A.; Zuo, F.; Woo, J.; Esser, M.T.; Tang, R.S. Adults 65 Years Old and Older Have Reduced Numbers of Functional Memory T Cells to Respiratory Syncytial Virus Fusion Protein. Clin. Vaccine Immunol. 2013, 20, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. MTOR Regulates Memory CD8 T-Cell Differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The MTOR Kinase Determines Effector versus Memory CD8+ T Cell Fate by Regulating the Expression of Transcription Factors T-Bet and Eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.; Bredfeldt, G.; Havsteen, B.; Lemke, H. Protein Kinase Activity of the Intracellular but Not of the Membrane-Associated Form of the KI-1 Antigen (CD30). Res. Immunol. 1990, 141, 13–31. [Google Scholar] [CrossRef]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global Burden of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Young Children: A Systematic Review and Meta-Analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Meissner, H.C.; Rennels, M.B.; Pickering, L.K.; Hall, C.B. Risk of Severe Respiratory Syncytial Virus Disease, Identification of High Risk Infants and Recommendations for Prophylaxis with Palivizumab [4]. Pediatr. Infect. Dis. J. 2004, 23, 284–285. [Google Scholar] [CrossRef]
- Chemaly, R.F.; Aitken, S.L.; Wolfe, C.R.; Jain, R.; Boeckh, M.J. Aerosolized Ribavirin: The Most Expensive Drug for Pneumonia. Transpl. Infect. Dis. 2016, 18, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, J.R.; Wilson, J.W.; Razonable, R.R. Oral Ribavirin Therapy for Respiratory Syncytial Virus Infections in Moderately to Severely Immunocompromised Patients. Transpl. Infect. Dis. 2014, 16, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, F.; Libre, C.; Oleinikov, A.; Tcherniuk, S. Chloroquine and pyrimethamine inhibit the replication of human respiratory syncytial virus A. J. Gen. Virol. 2021, 102, 001627. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Khare, A.; Oriss, T.B.; Raundhal, M.; Morse, C.; Yarlagadda, M.; Wenzel, S.E.; Moore, M.L.; Peebles, R.S.; Ray, A.; et al. Early Infection with Respiratory Syncytial Virus Impairs Regulatory T Cell Function and Increases Susceptibility to Allergic Asthma. Nat. Med. 2012, 18, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, X.; Yan, W.; Zeng, H.; Cheng, W. Qingfei oral liquid alleviates airway hyperresponsiveness and mucus hypersecretion via TRPV1 signaling in RSV-infected asthmatic mice. Biomed. Pharmacother. 2020, 128, 110340. [Google Scholar] [CrossRef]
- Yu, L.; Wang, J.; Zou, Y.; Zeng, H.; Cheng, W.; Jing, X. Qingfei oral liquid inhibited autophagy to alleviate inflammation via mTOR signaling pathway in RSV-infected asthmatic mice. Biomed. Pharmacother. 2021, 138, 111449. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luo, Y.; Wang, M.; Sun, L.; Huang, K.; Li, Y.; Chen, Y.; Ding, Y.; Zhang, X.; Jiao, L.; et al. Wuhu Decoction Regulates Dendritic Cell Autophagy in the Treatment of Respiratory Syncytial Virus (RSV)-Induced Mouse Asthma by AMPK/ULK1 Signaling Pathway. Med. Sci. Monit. 2019, 25, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Ren, K.; Lv, B.; Zhang, W.; Zhao, Y.; Tan, R.X.; Li, E. Baicalin from Scutellaria baicalensis blocks respiratory syncytial virus (RSV) infection and reduces inflammatory cell infiltration and lung injury in mice. Sci. Rep. 2016, 6, 35851. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Wu, Z.; Gao, B.; Xu, J. Analysis of Influence of Baicalin Joint Resveratrol Retention Enema on the TNF-α, SIgA, IL-2, IFN-γ of Rats with Respiratory Syncytial Virus Infection. Cell Biochem. Biophys. 2014, 70, 1305–1309. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Han, L.; Shi, X.L.; Wang, B.L.; Huang, H.; Wang, X.; Chen, D.F.; Ju, D.W.; Feng, M.Q. Baicalin Inhibits Autophagy Induced by Influenza A Virus H3N2. Antiviral Res. 2015, 113, 62–70. [Google Scholar] [CrossRef]
- Tian, Y.; Song, W.; Li, D.; Cai, L.; Zhao, Y. Resveratrol as a Natural Regulator of Autophagy for Prevention and Treatment of Cancer. Onco. Targets. Ther. 2019, 12, 8601–8609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Dong, H.; Jia, S.; Yang, Z. Resveratrol as a modulatory of apoptosis and autophagy in cancer therapy. Clin. Transl. Oncol. 2022, 24, 1219–1230. [Google Scholar] [CrossRef]
- Zang, N.; Xie, X.; Deng, Y.; Wu, S.; Wang, L.; Peng, C.; Li, S.; Ni, K.; Luo, Y.; Liu, E. Resveratrol-Mediated Gamma Interferon Reduction Prevents Airway Inflammation and Airway Hyperresponsiveness in Respiratory Syncytial Virus-Infected Immunocompromised Mice. J. Virol. 2011, 85, 13061–13068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, N.; Li, S.; Li, W.; Xie, X.; Ren, L.; Long, X.; Xie, J.; Deng, Y.; Fu, Z.; Xu, F.; et al. Resveratrol Suppresses Persistent Airway Inflammation and Hyperresponsivess Might Partially via Nerve Growth Factor in Respiratory Syncytial Virus-Infected Mice. Int. Immunopharmacol. 2015, 28, 121–128. [Google Scholar] [CrossRef]
- Xie, X.H.; Zang, N.; Li, S.; Wang, L.; Deng, Y.; He, Y.; Yang, X.; Liu, E. mei Resveratrol Inhibits Respiratory Syncytial Virus-Induced IL-6 Production, Decreases Viral Replication, and Downregulates TRIF Expression in Airway Epithelial Cells. Inflammation 2012, 35, 1392–1401. [Google Scholar] [CrossRef]
- Koch, A. Ausw: Berliner Klinische Wochenschrift, Band 19 (1882). Zent. Bakteriol. Mikrobiol. Hyg A Med. Mikrobiol. Infekt. Parasitol. 1982, 251, 287–296. [Google Scholar]
- Mbuh, T.P.; Ane-Anyangwe, I.; Adeline, W.; Pokam, B.D.T.; Meriki, H.D.; Mbacham, W.F. Bacteriologically confirmed extra pulmonary tuberculosis and treatment outcome of patients consulted and treated under program conditions in the littoral region of Cameroon. BMC Pulm. Med. 2019, 19, 17. [Google Scholar] [CrossRef]
- Lee, S.H. Tuberculosis Infection and Latent Tuberculosis. Tuberc. Respir. Dis. 2016, 79, 201–206. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021; pp. 1–57. [CrossRef]
- Kumar, R.; Singh, P.; Kolloli, A.; Shi, L.; Bushkin, Y.; Tyagi, S.; Subbian, S. Immunometabolism of Phagocytes During Mycobacterium tuberculosis Infection. Front. Mol. Biosci. 2019, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.M.; Ramakrishnan, L. The Role of the Granuloma in Expansion and Dissemination of Early Tuberculous Infection. Cell 2009, 136, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Blomgran, R.; Desvignes, L.; Briken, V.; Ernst, J.D. Mycobacterium Tuberculosis Inhibits Neutrophil Apoptosis, Leading to Delayed Activation of Naive CD4 T Cells. Cell Host Microbe 2012, 11, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Fratti, R.A.; Chua, J.; Vergne, I.; Deretic, V. Mycobacterium Tuberculosis Glycosylated Phosphatidylinositol Causes Phagosome Maturation Arrest. Proc. Natl. Acad. Sci. USA 2003, 100, 5437–5442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’arcy Hart, P.; Young, M.R.; Gordon, A.H.; Sullivan, K.H. Inhibition of Phagosome-Lysosome Fusion in Macrophages by Certain Mycobacteria Can Be Explained by Inhibition of Lysosomal Movements Observed after Phagocytosis. J. Exp. Med. 1987, 166, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J.M. Tuberculosis and M. Leprae Translocate from the Phagolysosome to the Cytosol in Myeloid Cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houben, D.; Demangel, C.; van Ingen, J.; Perez, J.; Baldeón, L.; Abdallah, A.M.; Caleechurn, L.; Bottai, D.; van Zon, M.; de Punder, K.; et al. ESX-1-Mediated Translocation to the Cytosol Controls Virulence of Mycobacteria. Cell. Microbiol. 2012, 14, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor CGAS Detects Mycobacterium Tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Mahla, R.S.; Kumar, A.; Tutill, H.J.; Krishnaji, S.T.; Sathyamoorthy, B.; Noursadeghi, M.; Breuer, J.; Pandey, A.K.; Kumar, H. NIX-mediated mitophagy regulate metabolic reprogramming in phagocytic cells during mycobacterial infection. Tuberculosis 2020, 126, 102046. [Google Scholar] [CrossRef]
- Sakowski, E.T.; Koster, S.; Celhay, C.P.; Park, H.S.; Shrestha, E.; Hetzenecker, S.E.; Maurer, K.; Cadwell, K.; Philips, J.A. Ubiquilin 1 Promotes IFN-γ-Induced Xenophagy of Mycobacterium tuberculosis. PLoS Pathog. 2015, 11, e1005076. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.H.; Nair, V.R.; Scharn, C.R.; Xavier, R.J.; Torrealba, J.R.; Shiloh, M.U.; Levine, B. Erratum: The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of Mycobacterium Tuberculosis and Anti-Tuberculous Host Defense (Cell Host & Microbe (2017) 21(1) (59–72) (S1931312816304772) (10.1016/j.Chom.2016.11.002)). Cell Host Microbe 2017, 22, 421–423. [Google Scholar] [CrossRef] [Green Version]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The Ubiquitin Ligase Parkin Mediates Resistance to Intracellular Pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Yi, M.; Chen, J.; Li, S.; Chen, W. Mycobacterium Tuberculosis EIS Gene Inhibits Macrophage Autophagy through Up-Regulation of IL-10 by Increasing the Acetylation of Histone H3. Biochem. Biophys. Res. Commun. 2016, 473, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Tagle, C.; Naves, R.; Balcells, M.E. Unraveling the Role of Micrornas in Mycobacterium Tuberculosis Infection and Disease: Advances and Pitfalls. Infect. Immun. 2020, 88, e00649-19. [Google Scholar] [CrossRef]
- Strong, E.J.; Jurcic Smith, K.L.; Saini, N.K.; Ng, T.W.; Porcelli, S.A.; Lee, S. Identification of Autophagy-Inhibiting Factors of Mycobacterium Tuberculosis by High-Throughput Loss-of-Function Screening. Infect. Immun. 2020, 88, e00269-20. [Google Scholar] [CrossRef]
- Romagnoli, A.; Etna, M.P.; Giacomini, E.; Pardini, M.; Remoli, M.E.; Corazzari, M.; Falasca, L.; Goletti, D.; Gafa, V.; Simeone, R.; et al. ESX-1 Dependent Impairment of Autophagic Flux by Mycobacterium Tuberculosis in Human Dendritic Cells. Autophagy 2012, 8, 1357–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V.; et al. Mycobacterium Tuberculosis Is Protected from NADPH Oxidase and LC3-Associated Phagocytosis by the LCP Protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720. [Google Scholar] [CrossRef] [Green Version]
- Dutta, N.K.; Bruiners, N.; Pinn, M.L.; Zimmerman, M.D.; Prideaux, B.; Dartois, V.; Gennaro, M.L.; Karakousis, P.C. Statin Adjunctive Therapy Shortens the Duration of TB Treatment in Mice. J. Antimicrob. Chemother. 2016, 71, 1570–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.W.; Gu, Y.; Peters, R.S.; Salgame, P.; Ellner, J.J.; Timmins, G.S.; Deretica, V. Ambroxol Induces Autophagy and Potentiates Rifampin Antimycobacterial Activity. Antimicrob. Agents Chemother. 2018, 62, e01019-18. [Google Scholar] [CrossRef] [Green Version]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.S.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as Adjunct Antituberculosis Therapy. Sci. Transl. Med. 2014, 6, 263ra159. [Google Scholar] [CrossRef]
- Sivangala Thandi, R.; Radhakrishnan, R.K.; Tripathi, D.; Paidipally, P.; Azad, A.K.; Schlesinger, L.S.; Samten, B.; Mulik, S.; Vankayalapati, R. Ornithine-A Urea Cycle Metabolite Enhances Autophagy and Controls Mycobacterium Tuberculosis Infection. Nat. Commun. 2020, 11, 3535. [Google Scholar] [CrossRef]
- Cerni, S.; Shafer, D.; To, K.; Venketaraman, V. Investigating the Role of Everolimus in MTOR Inhibition and Autophagy Promotion as a Potential Host-Directed Therapeutic Target in Mycobacterium Tuberculosis Infection. J. Clin. Med. 2019, 8, 232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Sun, J.; Wang, Y.; He, W.; Wang, L.; Zheng, Y.; Wu, J.; Zhang, Y.; Jiang, X. Antimycobacterial and Anti-Inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Mycobacterium Tuberculosis. Front. Microbiol. 2017, 8, 2142. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.; Zhao, D.; Shah, S.Z.A.; Sabir, N.; Wang, J.; Liao, Y.; Song, Y.; Dong, H.; Hussain Mangi, M.; Ni, J.; et al. Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells 2019, 8, 506. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wen, Z.; Liu, S.; Cai, Y.; Guo, J.; Xu, Y.; Lin, D.; Zhu, J.; Li, D.; Chen, X. Ibrutinib Suppresses Intracellular Mycobacterium Tuberculosis Growth by Inducing Macrophage Autophagy. J. Infect. 2020, 80, e19–e26. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Q.; Zhang, K.; Lin, D.; Feng, C.G.; Cai, Y.; Chen, X. Bazedoxifene Suppresses Intracellular Mycobacterium Tuberculosis Growth by Enhancing Autophagy. mSphere 2020, 5, e00124-20. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.M.; Andersson, B.; Lorell, C.; Raffetseder, J.; Larsson, M.; Blomgran, R. Autophagy Induction Targeting MTORC1 Enhances Mycobacterium Tuberculosis Replication in HIV Co-Infected Human Macrophages. Sci. Rep. 2016, 6, 28171. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, H.-M.; Kim, J.K.; Yang, C.-S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-α Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [Green Version]
- Mawatwal, S.; Behura, A.; Ghosh, A.; Kidwai, S.; Mishra, A.; Deep, A.; Agarwal, S.; Saha, S.; Singh, R.; Dhiman, R. Calcimycin Mediates Mycobacterial Killing by Inducing Intracellular Calcium-Regulated Autophagy in a P2RX7 Dependent Manner. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 3190–3200. [Google Scholar] [CrossRef]
- Mawatwal, S.; Behura, A.; Mishra, A.; Singh, R.; Dhiman, R. Calcimycin Induced IL-12 Production Inhibits Intracellular Mycobacterial Growth by Enhancing Autophagy. Cytokine 2018, 111, 1–12. [Google Scholar] [CrossRef]
- Mishra, A.; Behura, A.; Kumar, A.; Ghosh, A.; Naik, L.; Mawatwal, S.; Mohanty, S.S.; Mishra, A.; Saha, S.; Bhutia, S.K.; et al. Soybean Lectin Induces Autophagy through P2RX7 Dependent Activation of NF-ΚB-ROS Pathway to Kill Intracellular Mycobacteria. Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129806. [Google Scholar] [CrossRef]
- Ratjen, F.; Döring, G. Cystic Fibrosis. Lancet 2003, 361, 681–689. [Google Scholar] [CrossRef]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the Disease Liability of Variants in the Cystic Fibrosis Transmembrane Conductance Regulator Gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Antigny, F.; Norez, C.; Becq, F.; Vandebrouck, C. CFTR and Ca2+ Signaling in Cystic Fibrosis. Front. Pharmacol. 2011, 2, 67. [Google Scholar] [CrossRef] [Green Version]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-Dependent NLRP3 Activation Exacerbates the Pseudomonas Aeruginosa-Driven Inflammatory Response in Cystic Fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Rimessi, A.; Pozzato, C.; Carparelli, L.; Rossi, A.; Ranucci, S.; de Fino, I.; Cigana, C.; Talarico, A.; Wieckowski, M.R.; Ribeiro, C.M.P.; et al. Pharmacological Modulation of Mitochondrial Calcium Uniporter Controls Lung Inflammation in Cystic Fibrosis. Sci. Adv. 2020, 6, eaax9093. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR Induces Aggresome Formation and Lung Inflammation in Cystic Fibrosis through ROS-Mediated Autophagy Inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Villella, V.R.; Esposito, S.; Bruscia, E.M.; Vicinanza, M.; Cenci, S.; Guido, S.; Pettoello-Mantovani, M.; Carnuccio, R.; De Matteis, M.A.; Luini, A.; et al. Disease-Relevant Proteostasis Regulation of Cystic Fibrosis Transmembrane Conductance Regulator. Cell Death Differ. 2013, 20, 1101–1115. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, B.A.; Khweek, A.A.; Akhter, A.; Caution, K.; Kotrange, S.; Abdelaziz, D.H.A.; Newland, C.; Rosales-Reyes, R.; Kopp, B.; McCoy, K.; et al. Autophagy Stimulation by Rapamycin Suppresses Lung Inflammation and Infection by Burkholderia Cenocepacia in a Model of Cystic Fibrosis. Autophagy 2011, 7, 1359–1370. [Google Scholar] [CrossRef] [Green Version]
- Tazi, M.F.; Dakhlallah, D.A.; Caution, K.; Gerber, M.M.; Chang, S.W.; Khalil, H.; Kopp, B.T.; Ahmed, A.E.; Krause, K.; Davis, I.; et al. Elevated Mirc1/Mir17-92 Cluster Expression Negatively Regulates Autophagy and CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) Function in CF Macrophages. Autophagy 2016, 12, 2026–2037. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Patergnani, S.; Vitto, V.A.M.; Pinton, P.; Rimessi, A. Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front. Pharmacol. 2020, 11, 581114. [Google Scholar] [CrossRef]
- Del Porto, P.; Cifani, N.; Guarnieri, S.; Di Domenico, E.G.; Mariggiò, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional Cftr Alters the Bactericidal Activity of Human Macrophages against Pseudomonas Aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar] [CrossRef] [Green Version]
- Assani, K.; Tazi, M.F.; Amer, A.O.; Kopp, B.T. IFN-γ Stimulates Autophagy-Mediated Clearance of Burkholderia Cenocepacia in Human Cystic Fibrosis Macrophages. PLoS ONE 2014, 9, e96681. [Google Scholar] [CrossRef] [Green Version]
- Brockman, S.M.; Bodas, M.; Silverberg, D.; Sharma, A.; Vij, N. Dendrimer-Based Selective Autophagy-Induction Rescues ΔF508-CFTR and Inhibits Pseudomonas Aeruginosa Infection in Cystic Fibrosis. PLoS ONE 2017, 12, e0184793. [Google Scholar] [CrossRef]
- Ferrari, E.; Monzani, R.; Villella, V.R.; Esposito, S.; Saluzzo, F.; Rossin, F.; D’Eletto, M.; Tosco, A.; De Gregorio, F.; Izzo, V.; et al. Cysteamine Re-Establishes the Clearance of Pseudomonas Aeruginosa by Macrophages Bearing the Cystic Fibrosis-Relevant F508del-CFTR Mutation. Cell Death Dis. 2017, 8, e2544. [Google Scholar] [CrossRef] [Green Version]
- Caution, K.; Pan, A.; Krause, K.; Badr, A.; Hamilton, K.; Vaidya, A.; Gosu, H.; Daily, K.; Estfanous, S.; Gavrilin, M.A.; et al. Methylomic Correlates of Autophagy Activity in Cystic Fibrosis. J. Cyst. Fibros. 2019, 18, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Faraj, J.; Bodas, M.; Pehote, G.; Swanson, D.; Sharma, A.; Vij, N. Novel Cystamine-Core Dendrimer-Formulation Rescues ΔF508-CFTR and Inhibits Pseudomonas Aeruginosa Infection by Augmenting Autophagy. Expert Opin. Drug Deliv. 2019, 16, 177–186. [Google Scholar] [CrossRef]
- De Stefano, D.; Villella, V.R.; Esposito, S.; Tosco, A.; Sepe, A.; De Gregorio, F.; Salvadori, L.; Grassia, R.; Leone, C.A.; De Rosa, G.; et al. Restoration of CFTR Function in Patients with Cystic Fibrosis Carrying the F508del-CFTR Mutation. Autophagy 2014, 10, 2053–2074. [Google Scholar] [CrossRef] [Green Version]
- Devereux, G.; Fraser-Pitt, D.; Robertson, J.; Devlin, E.; Mercer, D.; O’Neil, D. Cysteamine as a Future Intervention in Cystic Fibrosis Against Current and Emerging Pathogens: A Patient-Based Ex Vivo Study Confirming Its Antimicrobial and Mucoactive Potential in Sputum. EBioMedicine 2015, 2, 1507–1512. [Google Scholar] [CrossRef] [Green Version]
- Devereux, G.; Steele, S.; Griffiths, K.; Devlin, E.; Fraser-Pitt, D.; Cotton, S.; Norrie, J.; Chrystyn, H.; O’Neil, D. An Open-Label Investigation of the Pharmacokinetics and Tolerability of Oral Cysteamine in Adults with Cystic Fibrosis. Clin. Drug Investig. 2016, 36, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Vu, C.B.; Bridges, R.J.; Pena-Rasgado, C.; Lacerda, A.E.; Bordwell, C.; Sewell, A.; Nichols, A.J.; Chandran, S.; Lonkar, P.; Picarella, D.; et al. Fatty Acid Cysteamine Conjugates as Novel and Potent Autophagy Activators That Enhance the Correction of Misfolded F508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). J. Med. Chem. 2017, 60, 458–473. [Google Scholar] [CrossRef]
- Emoto, C.; Fukuda, T.; Cox, S.; Christians, U.; Vinks, A.A. Development of a Physiologically-Based Pharmacokinetic Model for Sirolimus: Predicting Bioavailability Based on Intestinal CYP3A Content. CPT Pharmacometrics Syst. Pharmacol. 2013, 2, 59. [Google Scholar] [CrossRef]
- Bee, J.; Fuller, S.; Miller, S.; Johnson, S.R. Lung Function Response and Side Effects to Rapamycin for Lymphangioleiomyomatosis: A Prospective National Cohort Study. Thorax 2018, 73, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Haeri, A.; Osouli, M.; Bayat, F.; Alavi, S.; Dadashzadeh, S. Nanomedicine Approaches for Sirolimus Delivery: A Review of Pharmaceutical Properties and Preclinical Studies. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (MTOR)–Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a025924. [Google Scholar] [CrossRef] [Green Version]
- Mingione, A.; Ottaviano, E.; Barcella, M.; Merelli, I.; Rosso, L.; Armeni, T.; Cirilli, N.; Ghidoni, R.; Borghi, E.; Signorelli, P. Cystic Fibrosis Defective Response to Infection Involves Autophagy and Lipid Metabolism. Cells 2020, 9, 1845. [Google Scholar] [CrossRef]
- Zaman, K.; Carraro, S.; Doherty, J.; Henderson, E.M.; Lendermon, E.; Liu, L.; Verghese, G.; Zigler, M.; Ross, M.; Park, E.; et al. S-Nitrosylating Agents: A Novel Class of Compounds That Increase Cystic Fibrosis Transmembrane Conductance Regulator Expression and Maturation in Epithelial Cells. Mol. Pharmacol. 2006, 70, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Zaman, K.; Bennett, D.; Fraser-Butler, M.; Greenberg, Z.; Getsy, P.; Sattar, A.; Smith, L.; Corey, D.; Sun, F.; Hunt, J.; et al. S-Nitrosothiols Increases Cystic Fibrosis Transmembrane Regulator Expression and Maturation in the Cell Surface. Biochem. Biophys. Res. Commun. 2014, 443, 1257–1262. [Google Scholar] [CrossRef] [Green Version]
- Zaman, K.; Sawczak, V.; Zaidi, A.; Butler, M.; Bennett, D.; Getsy, P.; Zeinomar, M.; Greenberg, Z.; Forbes, M.; Rehman, S.; et al. Augmentation of Cftr Maturation by S-Nitrosoglutathione Reductase. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L263–L270. [Google Scholar] [CrossRef] [Green Version]
- Kim, V.; Criner, G.J. Chronic Bronchitis and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2013, 187, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Petrache, I.; Serban, K. Emphysema. Pathobiol. Hum. Dis. A Dyn. Encycl. Dis. Mech. 2014, 174, 2609–2624. [Google Scholar] [CrossRef]
- Polosukhin, V.V.; Richmond, B.W.; Du, R.H.; Cates, J.M.; Wu, P.; Nian, H.; Massion, P.P.; Ware, L.B.; Lee, J.W.; Kononov, A.V.; et al. Secretory IgA Deficiency in Individual Small Airways Is Associated with Persistent Inflammation and Remodeling. Am. J. Respir. Crit. Care Med. 2017, 195, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Patel, I.S.; Roberts, N.J.; Lloyd-Owen, S.J.; Sapsford, R.J.; Wedzicha, J.A. Airway Epithelial Inflammatory Responses and Clinical Parameters in COPD. Eur. Respir. J. 2003, 22, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of Inflammatory Cells in Airway Remodeling in COPD. Int. J. COPD 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, S.J.; Larivée, P.; Logun, C.; Angus, C.W.; Ognibene, F.P.; Shelhamer, J.H. Tumor Necrosis Factor-Alpha Induces Mucin Hypersecretion and MUC-2 Gene Expression by Human Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 1995, 12, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Fei, J.; Cao, P.; Zhang, C.; Tang, M.M.; Cheng, J.Y.; Zhao, H.; Fu, L. Serum Cadmium Positively Correlates with Inflammatory Cytokines in Patients with Chronic Obstructive Pulmonary Disease. Environ. Toxicol. 2022, 37, 151–160. [Google Scholar] [CrossRef]
- Terzikhan, N.; Verhamme, K.M.C.; Hofman, A.; Stricker, B.H.; Brusselle, G.G.; Lahousse, L. Prevalence and Incidence of COPD in Smokers and Non-Smokers: The Rotterdam Study. Eur. J. Epidemiol. 2016, 31, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Levine, B. Autophagy Balances Inflammation in Innate Immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Roscioli, E.; Hamon, R.; Lester, S.E.; Jersmann, H.P.A.; Reynolds, P.N.; Hodge, S. Airway Epithelial Cells Exposed to Wildfire Smoke Extract Exhibit Dysregulated Autophagy and Barrier Dysfunction Consistent with COPD. Respir. Res. 2018, 19, 234. [Google Scholar] [CrossRef]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 Regulates Autophagy in Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yu, G.; Yuan, S.; Tan, C.; Lian, P.; Fu, L.; Hou, Q.; Xu, B.; Wang, H. Cigarette Smoke-Induced Pulmonary Inflammation and Autophagy Are Attenuated in Ephx2-Deficient Mice. Inflammation 2017, 40, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; He, S.Y.; Lu, J.J.; Liu, C.; Lin, H.; Xu, C.Q.; Xie, L.H.; Sun, S.H. MicroRNA-21 Aggravates Chronic Obstructive Pulmonary Disease by Promoting Autophagy. Exp. Lung Res. 2018, 44, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.X.; Liu, S.S.; Li, K.; Cui, B.; Liu, C.; Hu, Z.W. Cigarette Smoke Promotes COPD by Activating Platelet-Activating Factor Receptor and Inducing Neutrophil Autophagic Death in Mice. Oncotarget 2017, 8, 74720–74735. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Shen, Y.; Wang, J.; Li, J.; Cao, J.; Zhang, J. Identification and Validation of Autophagy-Related Genes in Chronic Obstructive Pulmonary Disease. Int. J. COPD 2021, 16, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-Dependent Necroptosis Contributes to the Pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; White, A.; Wang, X.; Ko, J.; Choudhary, G.; Lange, T.; Rounds, S.; Lu, Q. Mitochondrial Fission Mediated Cigarette Smoke–Induced Pulmonary Endothelial Injury. Am. J. Respir. Cell Mol. Biol. 2020, 63, 637–651. [Google Scholar] [CrossRef]
- Wang, Q.; Unwalla, H.; Rahman, I. Dysregulation of Mitochondrial Complexes and Dynamics by Chronic Cigarette Smoke Exposure Utilizing MitoQC Reporter Mice. Mitochondrion 2022, 63, 43–50. [Google Scholar] [CrossRef]
- Wen, W.; Yu, G.; Liu, W.; Gu, L.; Chu, J.; Zhou, X.; Liu, Y.; Lai, G. Silencing FUNDC1 Alleviates Chronic Obstructive Pulmonary Disease by Inhibiting Mitochondrial Autophagy and Bronchial Epithelium Cell Apoptosis under Hypoxic Environment. J. Cell. Biochem. 2019, 120, 17602–17615. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, R.; Zhang, Y.; Shan, H.; Zhang, Q.; Yang, X.; Li, Y.; Zhang, J. Nix/BNIP3L-Dependent Mitophagy Accounts for Airway Epithelial Cell Injury Induced by Cigarette Smoke. J. Cell. Physiol. 2019, 234, 14210–14220. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy Promotes Primary Ciliogenesis by Removing OFD1 from Centriolar Satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Díaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional Interaction between Autophagy and Ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone Deacetylase 6-Mediated Selective Autophagy Regulates Copd-Associated Cilia Dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette Smoke-Induced Autophagy Impairment Accelerates Lung Aging, Copd-Emphysema Exacerbations and Pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient Autophagy Promotes Bronchial Epithelial Cell Senescence in Chronic Obstructive Pulmonary Disease. Oncoimmunology 2012, 1, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy Induction by SIRT6 through Attenuation of Insulin-like Growth Factor Signaling Is Involved in the Regulation of Human Bronchial Epithelial Cell Senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Song, J.H.; Ahn, J.H.; Lee, G.S.; Ahn, H.; Yoon, S.; Kang, S.G.; Kim, P.H.; Jeon, S.M.; Choi, E.J.; et al. Antiviral and Anti-Inflammatory Activity of Budesonide against Human Rhinovirus Infection Mediated via Autophagy Activation. Antiviral Res. 2018, 151, 87–96. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired Mitophagy Leads to Cigarette Smoke Stress-Induced Cellular Senescence: Implications for Chronic Obstructive Pulmonary Disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-Mediated Mitophagy Is Involved in Regulation of HBEC Senescence in COPD Pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-Regulated Mitophagy and Cellular Senescence during COPD Pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Pehote, G.; Silverberg, D.; Gulbins, E.; Vij, N. Autophagy Augmentation Alleviates Cigarette Smoke-Induced CFTR-Dysfunction, Ceramide-Accumulation and COPD-Emphysema Pathogenesis. Free Radic. Biol. Med. 2019, 131, 81–97. [Google Scholar] [CrossRef]
- Bodas, M.; Patel, N.; Silverberg, D.; Walworth, K.; Vij, N. Master Autophagy Regulator Transcription Factor EB Regulates Cigarette Smoke-Induced Autophagy Impairment and Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Antioxidants Redox Signal. 2017, 27, 150–167. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Duan, X.; Ji, R.; Perez, O.; Liu, C.; Merali, S. Cigarette Smoke Induces an Unfolded Protein Response in the Human Lung: A Proteomic Approach. Am. J. Respir. Cell Mol. Biol. 2008, 38, 541–550. [Google Scholar] [CrossRef]
- Kelsen, S.G. The Unfolded Protein Response in Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2016, 13, S138–S145. [Google Scholar] [CrossRef]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by P38 MAPK Couples ER Stress to Chaperone-Mediated Autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kuwano, K. Role of Chaperone-Mediated Autophagy in the Pathophysiology Including Pulmonary Disorders. Inflamm. Regen. 2021, 41, 29. [Google Scholar] [CrossRef]
- Hosaka, Y.; Araya, J.; Fujita, Y.; Kadota, T.; Tsubouchi, K.; Yoshida, M.; Minagawa, S.; Hara, H.; Kawamoto, H.; Watanabe, N.; et al. Chaperone-Mediated Autophagy Suppresses Apoptosis via Regulation of the Unfolded Protein Response during Chronic Obstructive Pulmonary Disease Pathogenesis. J. Immunol. 2020, 205, 1256–1267. [Google Scholar] [CrossRef]
- Li, D.; Hu, J.; Wang, T.; Zhang, X.; Liu, L.; Wang, H.; Wu, Y.; Xu, D.; Wen, F. Silymarin Attenuates Cigarette Smoke Extract-Induced Inflammation via Simultaneous Inhibition of Autophagy and ERK/P38 MAPK Pathway in Human Bronchial Epithelial Cells. Sci. Rep. 2016, 6, 37751. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Huang, J.; Dong, R.; Feng, Y.; Zhou, M. Therapeutic Potential of BLT1 Antagonist for COPD: Involvement of Inducing Autophagy and Ameliorating Inflammation. Drug Des. Devel. Ther. 2019, 13, 3105–3116. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Chen, J.; Huang, Q.; Zhan, Y.; Wang, T.; Wu, J.; Zhao, J.; Zeng, Z.; Lv, Y.; Xiao, C.; et al. MTMR14 Alleviates Chronic Obstructive Pulmonary Disease as a Regulator in Inflammation and Emphysema. Oxid. Med. Cell. Longev. 2022, 2022, 9300269. [Google Scholar] [CrossRef]
- Tran, I.; Ji, C.; Ni, I.; Min, T.; Tang, D.; Vij, N. Role of Cigarette Smoke-Induced Aggresome Formation in Chronic Obstructive Pulmonary Disease-Emphysema Pathogenesis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 159–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodas, M.; Van Westphal, C.; Carpenter-Thompson, R.; Mohanty, D.K.; Vij, N. Nicotine Exposure Induces Bronchial Epithelial Cell Apoptosis and Senescence via ROS Mediated Autophagy-Impairment. Free Radic. Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, G.; Yuan, S.; Tan, C.; Xie, J.; Ding, Y.; Lian, P.; Fu, L.; Hou, Q.; Xu, B.; et al. 14,15-Epoxyeicosatrienoic Acid Suppresses Cigarette Smoke Condensateinduced Inflammation in Lung Epithelial Cells by Inhibiting Autophagy. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2016, 311, L970–L980. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, H.; Sun, H.; Lu, R.; Zhang, C.; Gao, N.; Meng, Q.; Wu, S.; Wang, S.; Aschner, M.; et al. Erratum: Taurine Ameliorates Particulate Matter-Induced Emphysema by Switching on Mitochondrial NADH Dehydrogenase Genes (Proc. Natl. Acad. Sci. USA (2017) 114 (E9655–E9664). Doi: 10.1073/Pnas.1712465114). Proc. Natl. Acad. Sci. USA 2020, 117, 27062. [Google Scholar] [CrossRef] [Green Version]
- Son, E.S.; Kim, S.H.; Ryter, S.W.; Yeo, E.J.; Kyung, S.Y.; Kim, Y.J.; Jeong, S.H.; Lee, C.S.; Park, J.W. Quercetogetin Protects against Cigarette Smoke Extract-Induced Apoptosis in Epithelial Cells by Inhibiting Mitophagy. Toxicol. Vitr. 2018, 48, 170–178. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, W.; Wang, J.; Xie, Y.; Wang, W. Puerarin Inhibits FUNDCI-Mediated Mitochondrial Autophagy and CSE-Induced Apoptosis of Human Bronchial Epithelial Cells by Activating the PI3K/AKT/MTOR Signaling Pathway. Aging 2022, 14, 1253–1264. [Google Scholar] [CrossRef]
- Kyung, S.Y.; Kim, Y.J.; Son, E.S.; Jeong, S.H.; Park, J.W. The Phosphodiesterase 4 Inhibitor Roflumilast Protects against Cigarette Smoke Extract–Induced Mitophagy-Dependent Cell Death in Epithelial Cells. Tuberc. Respir. Dis. 2018, 81, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Carbone, M.; Adusumilli, P.S.; Alexander, H.R.; Baas, P.; Bardelli, F.; Bononi, A.; Bueno, R.; Felley-Bosco, E.; Galateau-Salle, F.; Jablons, D.; et al. Mesothelioma: Scientific Clues for Prevention, Diagnosis, and Therapy. CA Cancer J. Clin. 2019, 69, 402–429. [Google Scholar] [CrossRef] [Green Version]
- Qi, F.; Okimoto, G.; Jube, S.; Napolitano, A.; Pass, H.I.; Laczko, R.; Demay, R.M.; Khan, G.; Tiirikainen, M.; Rinaudo, C.; et al. Continuous Exposure to Chrysotile Asbestos Can Cause Transformation of Human Mesothelial Cells via HMGB1 and TNF-α Signaling. Am. J. Pathol. 2013, 183, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Patergnani, S.; Giorgi, C.; Suarez, J.; Goto, K.; Bononi, A.; Tanji, M.; Novelli, F.; Pastorino, S.; Xu, R.; et al. Asbestos Induces Mesothelial Cell Transformation via HMGB1-Driven Autophagy. Proc. Natl. Acad. Sci. USA 2020, 117, 25543–25552. [Google Scholar] [CrossRef]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of Autophagy Sensitizes Malignant Pleural Mesothelioma Cells to Dual PI3K/MTOR Inhibitors. Cell Death Dis. 2015, 6, e1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbone, D.; Follo, C.; Echeverry, N.; Gerbaudo, V.H.; Klabatsa, A.; Bueno, R.; Felley-Bosco, E.; Broaddus, V.C. Autophagy Correlates with the Therapeutic Responsiveness of Malignant Pleural Mesothelioma in 3D Models. PLoS ONE 2015, 10, e0134825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Liang, S.Q.; Yang, Z.; Yang, H.; Bruggmann, R.; Oberhaensli, S.; Berezowska, S.; Marti, T.M.; Hall, S.R.R.; Dorn, P.; et al. Malignant Pleural Mesothelioma Co-Opts BCL-XL and Autophagy to Escape Apoptosis. Cell Death Dis. 2021, 12, 406. [Google Scholar] [CrossRef] [PubMed]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Inhibition of Autophagy Initiation Potentiates Chemosensitivity in Mesothelioma. Mol. Carcinog. 2018, 57, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, S.H. Pro-Oxidant Activity of Sulforaphane and Cisplatin Potentiates Apoptosis and Simultaneously Promotes Autophagy in Malignant Mesothelioma Cells. Mol. Med. Rep. 2017, 16, 2133–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garva, R.; Thepmalee, C.; Yasamut, U.; Sudsaward, S.; Guazzelli, A.; Rajendran, R.; Tongmuang, N.; Kshunchai, S.; Meysami, P.; Limjindaporn, T.; et al. Sirtuin Family Members Selectively Regulate Autophagy in Osteosarcoma and Mesothelioma Cells in Response to Cellular Stress. Front. Oncol. 2019, 9, 949. [Google Scholar] [CrossRef]
- Carbone, M.; Gazdar, A.; Butel, J.S. SV40 and Human Mesothelioma. Transl. Lung Cancer Res. 2020, 9, S47–S59. [Google Scholar] [CrossRef]
- Novelli, F.; Bononi, A.; Wang, Q.; Bai, F.; Patergnani, S.; Kricek, F.; Haglund, E.; Suarez, J.S.; Tanji, M.; Xu, R.; et al. BAP1 Forms a Trimer with HMGB1 and HDAC1 That Modulates Gene × Environment Interaction with Asbestos. Proc. Natl. Acad. Sci. USA 2021, 118, e1757. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carinci, M.; Palumbo, L.; Pellielo, G.; Agyapong, E.D.; Morciano, G.; Patergnani, S.; Giorgi, C.; Pinton, P.; Rimessi, A. The Multifaceted Roles of Autophagy in Infectious, Obstructive, and Malignant Airway Diseases. Biomedicines 2022, 10, 1944. https://doi.org/10.3390/biomedicines10081944
Carinci M, Palumbo L, Pellielo G, Agyapong ED, Morciano G, Patergnani S, Giorgi C, Pinton P, Rimessi A. The Multifaceted Roles of Autophagy in Infectious, Obstructive, and Malignant Airway Diseases. Biomedicines. 2022; 10(8):1944. https://doi.org/10.3390/biomedicines10081944
Chicago/Turabian StyleCarinci, Marianna, Laura Palumbo, Giulia Pellielo, Esther Densu Agyapong, Giampaolo Morciano, Simone Patergnani, Carlotta Giorgi, Paolo Pinton, and Alessandro Rimessi. 2022. "The Multifaceted Roles of Autophagy in Infectious, Obstructive, and Malignant Airway Diseases" Biomedicines 10, no. 8: 1944. https://doi.org/10.3390/biomedicines10081944