
Chromosome Translocations, Gene Fusions, and Their Molecular Consequences in Pleomorphic Salivary Gland Adenomas †

 ,
,  ,
,  ,
,  ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Cytogenetic Landscape of PA
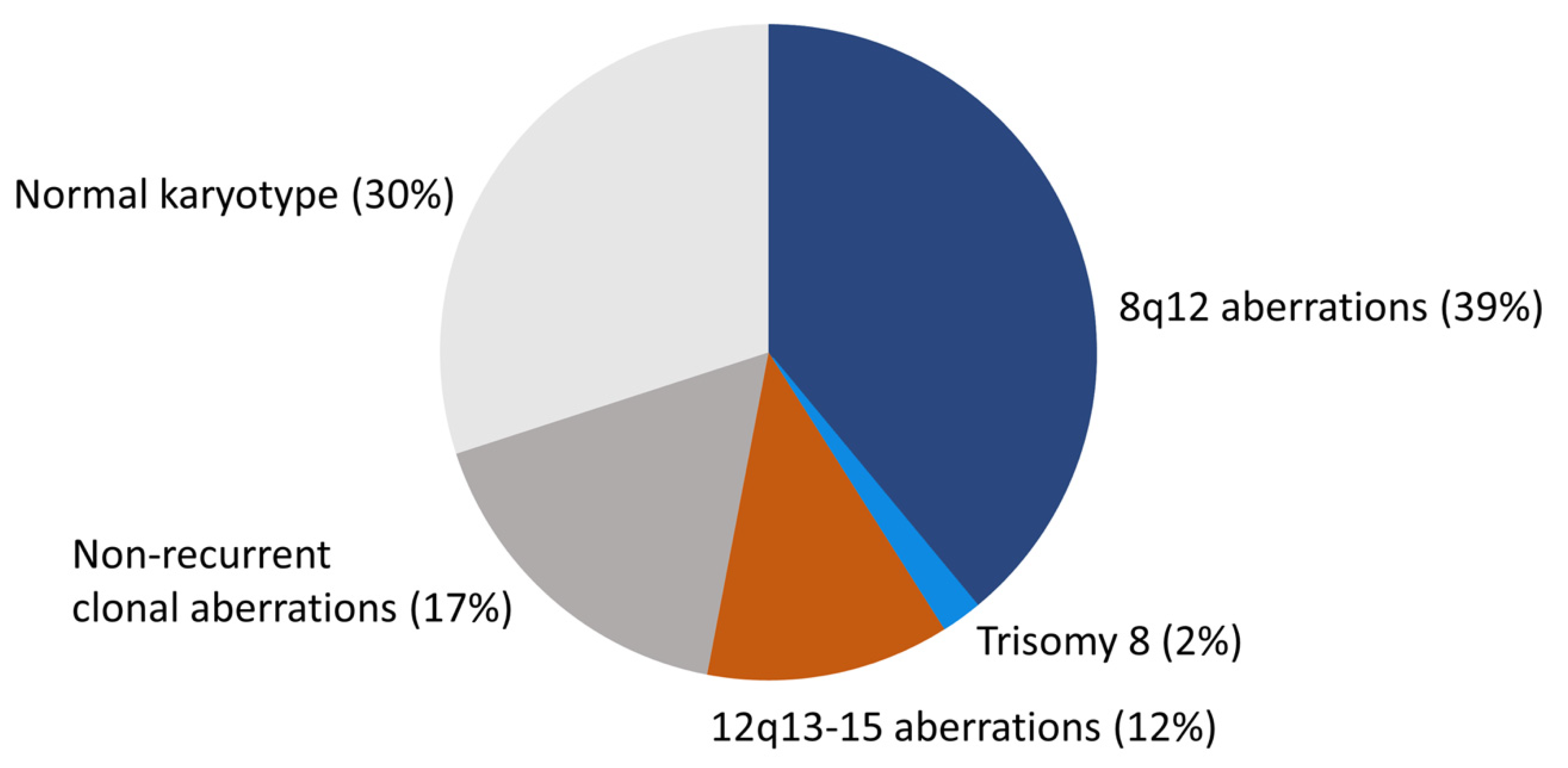
2.1. Overview of the Chromosomal Pattern in PA
2.2. Double Minute Chromosomes and Homogeneously Staining Regions
2.3. Ring Chromosomes and Dicentric Chromosomes
2.4. Polyclonal Aberrations in Radiation-Associated PAs
3. The Gene Fusion Landscape of PA
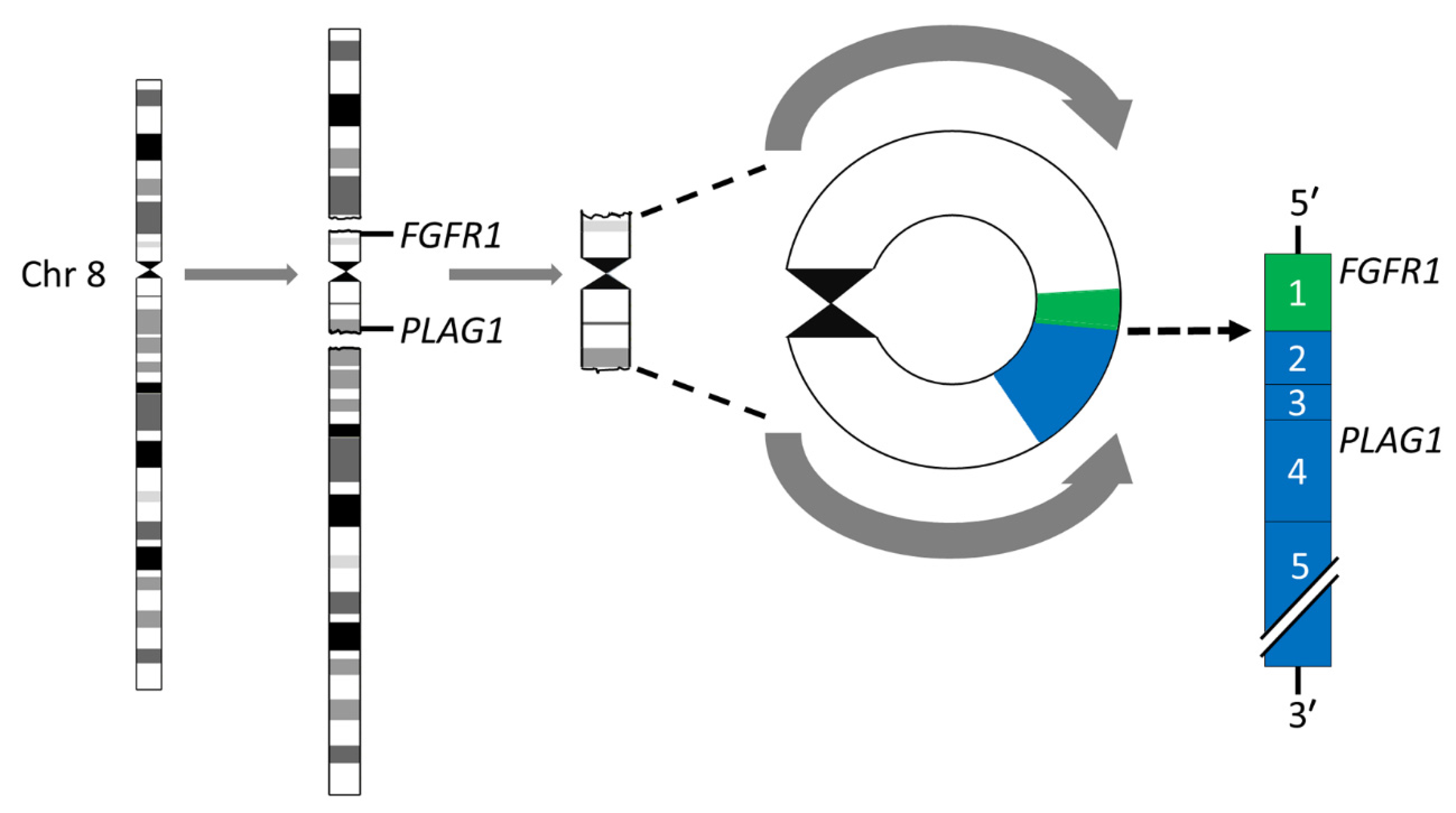
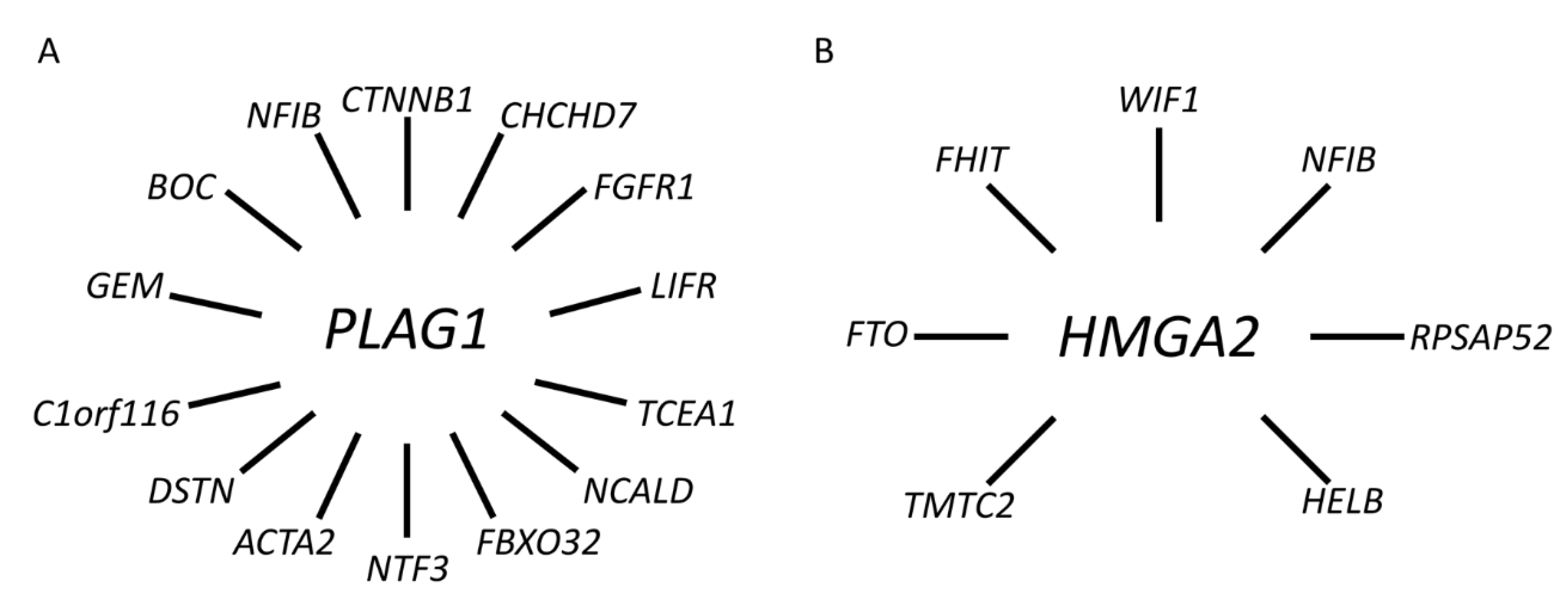
3.1. The Transcription Factor Gene PLAG1 Is the Target of Translocations and Rearrangements of 8q12 in PA
3.2. HMGA2 Is the Target Gene of Translocations and Rearrangements of 12q13-15 in PA
4. The 8q12 and 12q13-15 Rearrangements in PA Activate the HMGA2-PLAG1-IGF2 Pathway
5. Clinical Significance of Genomic Alterations in PA
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Classification of Tumours Editorial Board. International Agency for Research on Cancer, 5th ed.; WHO Classification of Tumours Series; Head and Neck Tumours [Internet; Beta Version Ahead of Print]; WHO Classification of Tumours Editorial Board: Lyon, France, 2022; Volume 9, Available online: https://tumourclassification.iarc.who.int/chapters/52 (accessed on 14 June 2022).
- Skalova, A.; Hyrcza, M.D.; Leivo, I. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Salivary Glands. Head Neck Pathol. 2022, 16, 40–53. [Google Scholar] [CrossRef]
- Andreasen, S.; Therkildsen, M.H.; Bjorndal, K.; Homoe, P. Pleomorphic adenoma of the parotid gland 1985-2010: A Danish nationwide study of incidence, recurrence rate, and malignant transformation. Head Neck 2016, 38 (Suppl. S1), E1364–E1369. [Google Scholar] [CrossRef]
- Valstar, M.H.; de Ridder, M.; van den Broek, E.C.; Stuiver, M.M.; van Dijk, B.A.C.; van Velthuysen, M.L.F.; Balm, A.J.M.; Smeele, L.E. Salivary gland pleomorphic adenoma in the Netherlands: A nationwide observational study of primary tumor incidence, malignant transformation, recurrence, and risk factors for recurrence. Oral Oncol. 2017, 66, 93–99. [Google Scholar] [CrossRef]
- Mark, J.; Ekedahl, C. Polyclonal chromosomal evolution in a benign mixed salivary gland tumor. Cancer Genet. Cytogenet. 1987, 28, 237–243. [Google Scholar] [CrossRef]
- Enlund, F.; Nordkvist, A.; Sahlin, P.; Mark, J.; Stenman, G. Expression of PLAG1 and HMGIC proteins and fusion transcripts in radiation-associated pleomorphic adenomas. Int. J. Oncol. 2002, 20, 713–716. [Google Scholar] [CrossRef]
- Hernandez-Prera, J.C.; Faquin, W.C.; Ihrler, S.; Katabi, N.; Weinreb, I.; Altemani, A.; Machado de Sousa, S.O.; Wasserman, J.K. Pleomorphic Adenoma. In WHO Classification of Tumours Editorial Board, 5th ed.; WHO Classification of Tumours Series; Head and Neck Tumours [Internet; Beta Version Ahead of Print]; International Agency for Research on Cancer: Lyon, France, 2022; Volume 9, Available online: https://tumourclassification.iarc.who.int/chapters/52 (accessed on 14 June 2022).
- Hernandez-Prera, J.C.; Skalova, A.; Franchi, A.; Rinaldo, A.; Vander Poorten, V.; Zbaren, P.; Ferlito, A.; Wenig, B.M. Pleomorphic adenoma: The great mimicker of malignancy. Histopathology 2021, 79, 279–290. [Google Scholar] [CrossRef]
- Katabi, N.; Chiosea, S.; Fonseca, I.; Ihrler, S.; Klijanienko, J.; Altemani, A. Carcinoma ex Pleomorphic Adenoma. In WHO Classification of Tumours Editorial Board, 5th ed.; WHO Classification of Tumours Series; Head and Neck Tumours [Internet; Beta Version Ahead of Print]; International Agency for Research on Cancer: Lyon, France, 2022; Volume 9, Available online: https://tumourclassification.iarc.who.int/chapters/52 (accessed on 14 June 2022).
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. The mixed salivary gland tumor—A normally benign human neoplasm frequently showing specific chromosomal abnormalities. Cancer Genet. Cytogenet. 1980, 2, 231–241. [Google Scholar] [CrossRef]
- Kas, K.; Voz, M.L.; Röijer, E.; Aström, A.K.; Meyen, E.; Stenman, G.; Van de Ven, W.J. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat. Genet. 1997, 15, 170–174. [Google Scholar] [CrossRef]
- Geurts, J.M.; Schoenmakers, E.F.; Röijer, E.; Stenman, G.; Van de Ven, W.J. Expression of reciprocal hybrid transcripts of HMGIC and FHIT in a pleomorphic adenoma of the parotid gland. Cancer Res. 1997, 57, 13–17. [Google Scholar]
- Geurts, J.M.; Schoenmakers, E.F.; Röijer, E.; Aström, A.K.; Stenman, G.; van de Ven, W.J. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene 1998, 16, 865–872. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C.; Stenman, G. Chromosomal patterns in a benign human neoplasm, the mixed salivary gland tumour. Hereditas 1982, 96, 141–148. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Stenman, G.; Mark, J. Specificity of the involvement of chromosomes 8 and 12 in human mixed salivary-gland tumours. J. Oral. Pathol. 1983, 12, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Voz, M.L.; Aström, A.K.; Kas, K.; Mark, J.; Stenman, G.; Van de Ven, W.J. The recurrent translocation t(5;8)(p13;q12) in pleomorphic adenomas results in upregulation of PLAG1 gene expression under control of the LIFR promoter. Oncogene 1998, 16, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Stenman, G. Fusion oncogenes in salivary gland tumors: Molecular and clinical consequences. Head Neck Pathol. 2013, 7 (Suppl. S1), S12–S19. [Google Scholar] [CrossRef]
- Astrom, A.K.; Voz, M.L.; Kas, K.; Roijer, E.; Wedell, B.; Mandahl, N.; Van de Ven, W.; Mark, J.; Stenman, G. Conserved mechanism of PLAG1 activation in salivary gland tumors with and without chromosome 8q12 abnormalities: Identification of SII as a new fusion partner gene. Cancer Res. 1999, 59, 918–923. [Google Scholar] [PubMed]
- Skalova, A.; Hyrcza, M.D.; Vanecek, T.; Baneckova, M.; Leivo, I. Fusion-positive salivary gland carcinomas. Genes Chromosomes Cancer 2022, 61, 228–243. [Google Scholar] [CrossRef]
- Andersson, M.K.; Stenman, G. The landscape of gene fusions and somatic mutations in salivary gland neoplasms—Implications for diagnosis and therapy. Oral Oncol. 2016, 57, 63–69. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C. Cytogenetics of the human benign mixed salivary gland tumour. Hereditas 1983, 99, 115–129. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R. Cytogenetical observations in 100 human benign pleomorphic adenomas: Specificity of the chromosomal aberrations and their relationship to sites of localized oncogenes. Anticancer Res. 1986, 6, 299–308. [Google Scholar]
- Mark, J.; Sandros, J.; Wedell, B.; Stenman, G.; Ekedahl, C. Significance of the choice of tissue culture technique on the chromosomal patterns in human mixed salivary gland tumors. Cancer Genet. Cytogenet. 1988, 33, 229–244. [Google Scholar] [CrossRef]
- Sandros, J.; Stenman, G.; Mark, J. Cytogenetic and molecular observations in human and experimental salivary gland tumors. Cancer Genet. Cytogenet. 1990, 44, 153–167. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Wedell, B. Impact of the in vitro technique used on the cytogenetic patterns in pleomorphic adenomas. Cancer Genet. Cytogenet. 1997, 95, 9–15. [Google Scholar] [CrossRef]
- Stenman, G. Fusion oncogenes and tumor type specificity—Insights from salivary gland tumors. Semin. Cancer Biol. 2005, 15, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Bullerdiek, J.; Bartnitzke, S.; Weinberg, M.; Chilla, R.; Haubrich, J.; Schloot, W. Rearrangements of chromosome region 12q13----q15 in pleomorphic adenomas of the human salivary gland (PSA). Cytogenet Cell Genet. 1987, 45, 187–190. [Google Scholar] [CrossRef]
- Bullerdiek, J.; Boschen, C.; Bartnitzke, S. Aberrations of chromosome 8 in mixed salivary gland tumors--cytogenetic findings on seven cases. Cancer Genet. Cytogenet. 1987, 24, 205–212. [Google Scholar] [CrossRef]
- Bullerdiek, J.; Chilla, R.; Haubrich, J.; Meyer, K.; Bartnitzke, S. A causal relationship between chromosomal rearrangements and the genesis of salivary gland pleomorphic adenomas. Arch. Otorhinolaryngol. 1988, 245, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Bullerdiek, J.; Wobst, G.; Meyer-Bolte, K.; Chilla, R.; Haubrich, J.; Thode, B.; Bartnitzke, S. Cytogenetic subtyping of 220 salivary gland pleomorphic adenomas: Correlation to occurrence, histological subtype, and in vitro cellular behavior. Cancer Genet. Cytogenet. 1993, 65, 27–31. [Google Scholar] [CrossRef]
- Martins, C.; Fonseca, I.; Felix, A.; Roque, L.; Soares, J. Benign salivary gland tumors: A cytogenetic study of 21 cases. J. Surg. Oncol. 1995, 60, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Hrynchak, M.; White, V.; Berean, K.; Horsman, D. Cytogenetic findings in seven lacrimal gland neoplasms. Cancer Genet. Cytogenet. 1994, 75, 133–138. [Google Scholar] [CrossRef]
- Mark, H.F.; Hanna, I.; Gnepp, D.R. Cytogenetic analysis of salivary gland type tumors. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1996, 82, 187–192. [Google Scholar] [CrossRef]
- Jin, C.; Martins, C.; Jin, Y.; Wiegant, J.; Wennerberg, J.; Dictor, M.; Gisselsson, D.; Strombeck, B.; Fonseca, I.; Mitelman, F.; et al. Characterization of chromosome aberrations in salivary gland tumors by FISH, including multicolor COBRA-FISH. Genes Chromosomes Cancer 2001, 30, 161–167. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. (Eds.) Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer: 2022. Available online: https://mitelmandatabase.isb-cgc.org (accessed on 21 June 2022).
- Afshari, M.K.; Fehr, A.; Nevado, P.T.; Andersson, M.K.; Stenman, G. Activation of PLAG1 and HMGA2 by gene fusions involving the transcriptional regulator gene NFIB. Genes Chromosomes Cancer 2020, 59, 652–660. [Google Scholar] [CrossRef]
- Asp, J.; Persson, F.; Kost-Alimova, M.; Stenman, G. CHCHD7-PLAG1 and TCEA1-PLAG1 gene fusions resulting from cryptic, intrachromosomal 8q rearrangements in pleomorphic salivary gland adenomas. Genes Chromosomes Cancer 2006, 45, 820–828. [Google Scholar] [CrossRef]
- Persson, F.; Winnes, M.; Andrén, Y.; Wedell, B.; Dahlenfors, R.; Asp, J.; Mark, J.; Enlund, F.; Stenman, G. High-resolution array CGH analysis of salivary gland tumors reveals fusion and amplification of the FGFR1 and PLAG1 genes in ring chromosomes. Oncogene 2008, 27, 3072–3080. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C. On double-minutes and their origin in a benign human neoplasm, a mixed salivary gland tumour. Anticancer Res. 1982, 2, 261–264. [Google Scholar] [PubMed]
- Di Palma, S.; Lambros, M.B.; Savage, K.; Jones, C.; Mackay, A.; Dexter, T.; Iravani, M.; Fenwick, K.; Ashworth, A.; Reis-Filho, J.S. Oncocytic change in pleomorphic adenoma: Molecular evidence in support of an origin in neoplastic cells. J. Clin. Pathol. 2007, 60, 492–499. [Google Scholar] [CrossRef]
- Persson, F.; Andrén, Y.; Winnes, M.; Wedell, B.; Nordkvist, A.; Gudnadottir, G.; Dahlenfors, R.; Sjögren, H.; Mark, J.; Stenman, G. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes Chromosomes Cancer 2009, 48, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Roijer, E.; Nordkvist, A.; Strom, A.K.; Ryd, W.; Behrendt, M.; Bullerdiek, J.; Mark, J.; Stenman, G. Translocation, deletion/amplification, and expression of HMGIC and MDM2 in a carcinoma ex pleomorphic adenoma. Am. J. Pathol. 2002, 160, 433–440. [Google Scholar] [CrossRef]
- Rao, P.H.; Murty, V.V.; Louie, D.C.; Chaganti, R.S. Nonsyntenic amplification of MYC with CDK4 and MDM2 in a malignant mixed tumor of salivary gland. Cancer Genet. Cytogenet. 1998, 105, 160–163. [Google Scholar] [CrossRef]
- McClintock, B. The Production of Homozygous Deficient Tissues with Mutant Characteristics by Means of the Aberrant Mitotic Behavior of Ring-Shaped Chromosomes. Genetics 1938, 23, 315–376. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Pettersson, L.; Hoglund, M.; Heidenblad, M.; Gorunova, L.; Wiegant, J.; Mertens, F.; Dal Cin, P.; Mitelman, F.; Mandahl, N. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. USA 2000, 97, 5357–5362. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Ekedahl, C. Specificity and implications of ring chromosomes and dicentrics in benign mixed salivary gland tumours. Acta Pathol. Microbiol. Immunol. Scand. A 1983, 91, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Dalin, M.G.; Katabi, N.; Persson, M.; Lee, K.W.; Makarov, V.; Desrichard, A.; Walsh, L.A.; West, L.; Nadeem, Z.; Ramaswami, D.; et al. Multi-dimensional genomic analysis of myoepithelial carcinoma identifies prevalent oncogenic gene fusions. Nat. Commun. 2017, 8, 1197. [Google Scholar] [CrossRef]
- Mark, J.; Dahlenfors, R.; Wedell, B. Is pleomorphic adenoma of the salivary glands a tumor of congenital or very early origin? Oncol. Rep. 1996, 3, 1075–1077. [Google Scholar] [CrossRef]
- Kas, K.; Voz, M.L.; Hensen, K.; Meyen, E.; Van de Ven, W.J. Transcriptional activation capacity of the novel PLAG family of zinc finger proteins. J. Biol. Chem. 1998, 273, 23026–23032. [Google Scholar] [CrossRef]
- Van Dyck, F.; Declercq, J.; Braem, C.V.; Van de Ven, W.J. PLAG1, the prototype of the PLAG gene family: Versatility in tumour development (review). Int. J. Oncol. 2007, 30, 765–774. [Google Scholar] [CrossRef]
- Juma, A.R.; Damdimopoulou, P.E.; Grommen, S.V.; Van de Ven, W.J.; De Groef, B. Emerging role of PLAG1 as a regulator of growth and reproduction. J. Endocrinol. 2016, 228, R45–R56. [Google Scholar] [CrossRef]
- Hensen, K.; Braem, C.; Declercq, J.; Van Dyck, F.; Dewerchin, M.; Fiette, L.; Denef, C.; Van de Ven, W.J. Targeted disruption of the murine Plag1 proto-oncogene causes growth retardation and reduced fertility. Dev. Growth Differ. 2004, 46, 459–470. [Google Scholar] [CrossRef]
- Voz, M.L.; Agten, N.S.; Van de Ven, W.J.; Kas, K. PLAG1, the main translocation target in pleomorphic adenoma of the salivary glands, is a positive regulator of IGF-II. Cancer Res. 2000, 60, 106–113. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S. Mutation Hotspots in the beta-Catenin Gene: Lessons from the Human Cancer Genome Databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef]
- Persson, M.; Andren, Y.; Moskaluk, C.A.; Frierson, H.F., Jr.; Cooke, S.L.; Futreal, P.A.; Kling, T.; Nelander, S.; Nordkvist, A.; Persson, F.; et al. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes Chromosomes Cancer 2012, 51, 805–817. [Google Scholar] [CrossRef]
- Bubola, J.; MacMillan, C.M.; Demicco, E.G.; Chami, R.A.; Chung, C.T.; Leong, I.; Marrano, P.; Onkal, Z.; Swanson, D.; Veremis, B.M.; et al. Targeted RNA sequencing in the routine clinical detection of fusion genes in salivary gland tumors. Genes Chromosomes Cancer 2021, 60, 695–708. [Google Scholar] [CrossRef]
- Baněčková, M.; Uro-Coste, E.; Ptáková, N.; Šteiner, P.; Stanowska, O.; Benincasa, G.; Colella, G.; Vondrák, J., Jr.; Michal, M.; Leivo, I.; et al. What is hiding behind S100 protein and SOX10 positive oncocytomas? Oncocytic pleomorphic adenoma and myoepithelioma with novel gene fusions in a subset of cases. Hum. Pathol. 2020, 103, 52–62. [Google Scholar] [CrossRef]
- Pei, J.; Liu, J.C.; Ehya, H.; Wei, S. BOC-PLAG1, a new fusion gene of pleomorphic adenoma: Identified in a fine-needle aspirate by RNA next-generation sequencing. Diagn. Cytopathol. 2021, 49, 790–792. [Google Scholar] [CrossRef]
- Ashar, H.R.; Fejzo, M.S.; Tkachenko, A.; Zhou, X.; Fletcher, J.A.; Weremowicz, S.; Morton, C.C.; Chada, K. Disruption of the architectural factor HMGI-C: DNA-binding AT hook motifs fused in lipomas to distinct transcriptional regulatory domains. Cell 1995, 82, 57–65. [Google Scholar] [CrossRef]
- Tkachenko, A.; Ashar, H.R.; Meloni, A.M.; Sandberg, A.A.; Chada, K.K. Misexpression of disrupted HMGI architectural factors activates alternative pathways of tumorigenesis. Cancer Res. 1997, 57, 2276–2280. [Google Scholar]
- Mansoori, B.; Mohammadi, A.; Ditzel, H.J.; Duijf, P.H.G.; Khaze, V.; Gjerstorff, M.F.; Baradaran, B. HMGA2 as a Critical Regulator in Cancer Development. Genes 2021, 12, 269. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Fusco, A.; Esposito, F. HMGA and Cancer: A Review on Patent Literatures. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Tessari, M.A.; Gostissa, M.; Altamura, S.; Sgarra, R.; Rustighi, A.; Salvagno, C.; Caretti, G.; Imbriano, C.; Mantovani, R.; Del Sal, G.; et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol. Cell. Biol. 2003, 23, 9104–9116. [Google Scholar] [CrossRef]
- Klemke, M.; Muller, M.H.; Wosniok, W.; Markowski, D.N.; Nimzyk, R.; Helmke, B.M.; Bullerdiek, J. Correlated expression of HMGA2 and PLAG1 in thyroid tumors, uterine leiomyomas and experimental models. PLoS ONE 2014, 9, e88126. [Google Scholar] [CrossRef]
- Andersson, M.K.; Aman, P.; Stenman, G. IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gen.ne-Driven Tumors. Cells 2019, 8, 913. [Google Scholar] [CrossRef]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef]
- Sun, L.; Petrone, J.S.; McNulty, S.N.; Evenson, M.J.; Zhu, X.; Robinson, J.A.; Chernock, R.D.; Duncavage, E.J.; Pfeifer, J.D. Comparison of gene fusion detection methods in salivary gland tumors. Hum. Pathol. 2022, 123, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A.; Ihrler, S.; Baněčková, M.; Costés Martineau, V.; Mantsopoulos, K.; Hartmann, A.; Iro, H.; Stoehr, R.; Skálová, A. HMGA2-WIF1 Rearrangements Characterize a Distinctive Subset of Salivary Pleomorphic Adenomas With Prominent Trabecular (Canalicular Adenoma-like) Morphology. Am. J. Surg. Pathol. 2022, 46, 190–199. [Google Scholar] [CrossRef]
- Wasserman, J.K.; Dickson, B.C.; Smith, A.; Swanson, D.; Purgina, B.M.; Weinreb, I. Metastasizing Pleomorphic Adenoma: Recurrent PLAG1/HMGA2 Rearrangements and Identification of a Novel HMGA2-TMTC2 Fusion. Am. J. Surg. Pathol. 2019, 43, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Queimado, L.; Lopes, C.S.; Reis, A.M. WIF1, an inhibitor of the Wnt pathway, is rearranged in salivary gland tumors. Genes Chromosomes Cancer 2007, 46, 215–225. [Google Scholar] [CrossRef]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Abi Habib, W.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; Le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef]
- Gicquel, C.; Rossignol, S.; Cabrol, S.; Houang, M.; Steunou, V.; Barbu, V.; Danton, F.; Thibaud, N.; Le Merrer, M.; Burglen, L.; et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet. 2005, 37, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Azzi, S.; Abi Habib, W.; Netchine, I. Beckwith-Wiedemann and Russell-Silver Syndromes: From new molecular insights to the comprehension of imprinting regulation. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 30–38. [Google Scholar] [CrossRef]
- Leszinski, G.S.; Warncke, K.; Hoefele, J.; Wagner, M. A case report and review of the literature indicate that HMGA2 should be added as a disease gene for Silver-Russell syndrome. Gene 2018, 663, 110–114. [Google Scholar] [CrossRef]
- Vado, Y.; Pereda, A.; Llano-Rivas, I.; Gorria-Redondo, N.; Diez, I.; Perez de Nanclares, G. Novel Variant in PLAG1 in a Familial Case with Silver-Russell Syndrome Suspicion. Genes 2020, 11, 1461. [Google Scholar] [CrossRef]
- Psychogios, G.; Bohr, C.; Constantinidis, J.; Canis, M.; Vander Poorten, V.; Plzak, J.; Knopf, A.; Betz, C.; Guntinas-Lichius, O.; Zenk, J. Review of surgical techniques and guide for decision making in the treatment of benign parotid tumors. Eur. Arch. Otorhinolaryngol. 2021, 278, 15–29. [Google Scholar] [CrossRef]
- Rooker, S.A.; Van Abel, K.M.; Yin, L.X.; Nagelschneider, A.A.; Price, D.L.; Olsen, K.D.; Janus, J.R.; Kasperbauer, J.L.; Moore, E.J. Risk factors for subsequent recurrence after surgical treatment of recurrent pleomorphic adenoma of the parotid gland. Head Neck 2021, 43, 1088–1096. [Google Scholar] [CrossRef]
- Alzumaili, B.; Xu, B.; Saliba, M.; Abuhashem, A.; Ganly, I.; Ghossein, R.; Katabi, N. Clinicopathologic Characteristics and Prognostic Factors of Primary and Recurrent Pleomorphic Adenoma: A Single Institution Retrospective Study of 705 Cases. Am. J. Surg. Pathol. 2022, 46, 854–862. [Google Scholar] [CrossRef] [PubMed]
- von Holstein, S.L.; Fehr, A.; Persson, M.; Nickelsen, M.; Therkildsen, M.H.; Prause, J.U.; Heegaard, S.; Stenman, G. Lacrimal gland pleomorphic adenoma and carcinoma ex pleomorphic adenoma: Genomic profiles, gene fusions, and clinical characteristics. Ophthalmology 2014, 121, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Asahina, M.; Saito, T.; Hayashi, T.; Fukumura, Y.; Mitani, K.; Yao, T. Clinicopathological effect of PLAG1 fusion genes in pleomorphic adenoma and carcinoma ex pleomorphic adenoma with special emphasis on histological features. Histopathology 2019, 74, 514–525. [Google Scholar] [CrossRef]
- Katabi, N.; Xu, B.; Jungbluth, A.A.; Zhang, L.; Shao, S.Y.; Lane, J.; Ghossein, R.; Antonescu, C.R. PLAG1 immunohistochemistry is a sensitive marker for pleomorphic adenoma: A comparative study with PLAG1 genetic abnormalities. Histopathology 2018, 72, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Dalton, J.D.; Shivakumar, B.; Krane, J.F. PLAG1 alteration in carcinoma ex pleomorphic adenoma: Immunohistochemical and fluorescence in situ hybridization studies of 22 cases. Head Neck Pathol. 2012, 6, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Rotellini, M.; Palomba, A.; Baroni, G.; Franchi, A. Diagnostic utility of PLAG1 immunohistochemical determination in salivary gland tumors. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Hisaoka, M.; Nagao, Y.; Hashimoto, H. Aberrant PLAG1 expression in pleomorphic adenomas of the salivary gland: A molecular genetic and immunohistochemical study. Virchows Arch. Int. J. Pathol. 2011, 458, 583–592. [Google Scholar] [CrossRef]
- Katabi, N.; Ghossein, R.; Ho, A.; Dogan, S.; Zhang, L.; Sung, Y.S.; Antonescu, C.R. Consistent PLAG1 and HMGA2 abnormalities distinguish carcinoma ex-pleomorphic adenoma from its de novo counterparts. Hum. Pathol. 2015, 46, 26–33. [Google Scholar] [CrossRef]
- Pareja, F.; Da Cruz Paula, A.; Gularte-Mérida, R.; Vahdatinia, M.; Li, A.; Geyer, F.C.; da Silva, E.M.; Nanjangud, G.; Wen, H.Y.; Varga, Z.; et al. Pleomorphic adenomas and mucoepidermoid carcinomas of the breast are underpinned by fusion genes. NPJ Breast Cancer 2020, 6, 20. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stenman, G.; Fehr, A.; Skálová, A.; Vander Poorten, V.; Hellquist, H.; Mikkelsen, L.H.; Saba, N.F.; Guntinas-Lichius, O.; Chiesa-Estomba, C.M.; Andersson, M.K.; et al. Chromosome Translocations, Gene Fusions, and Their Molecular Consequences in Pleomorphic Salivary Gland Adenomas. Biomedicines 2022, 10, 1970. https://doi.org/10.3390/biomedicines10081970
Stenman G, Fehr A, Skálová A, Vander Poorten V, Hellquist H, Mikkelsen LH, Saba NF, Guntinas-Lichius O, Chiesa-Estomba CM, Andersson MK, et al. Chromosome Translocations, Gene Fusions, and Their Molecular Consequences in Pleomorphic Salivary Gland Adenomas. Biomedicines. 2022; 10(8):1970. https://doi.org/10.3390/biomedicines10081970
Chicago/Turabian StyleStenman, Göran, Andre Fehr, Alena Skálová, Vincent Vander Poorten, Henrik Hellquist, Lauge Hjorth Mikkelsen, Nabil F. Saba, Orlando Guntinas-Lichius, Carlos Miguel Chiesa-Estomba, Mattias K. Andersson, and et al. 2022. "Chromosome Translocations, Gene Fusions, and Their Molecular Consequences in Pleomorphic Salivary Gland Adenomas" Biomedicines 10, no. 8: 1970. https://doi.org/10.3390/biomedicines10081970
APA StyleStenman, G., Fehr, A., Skálová, A., Vander Poorten, V., Hellquist, H., Mikkelsen, L. H., Saba, N. F., Guntinas-Lichius, O., Chiesa-Estomba, C. M., Andersson, M. K., & Ferlito, A. (2022). Chromosome Translocations, Gene Fusions, and Their Molecular Consequences in Pleomorphic Salivary Gland Adenomas. Biomedicines, 10(8), 1970. https://doi.org/10.3390/biomedicines10081970