
The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Design of the Study
- -
- Death;
- -
- Lung transplantation;
- -
- Interruption of the treatment due to any cause.
- Patients were excluded from the study in case of:
- -
- Inability or refusal to provide informed consent to participate in clinical studies;
- -
- Less than one month of antifibrotic treatment;
- -
- Previous antifibrotic treatment at baseline.
2.2. Lung Function Tests
2.3. Statistical Analysis
3. Results
3.1. Study Population
3.2. Subgroup Analysis: FPF
3.3. Subgroup Analysis: PF-ILD
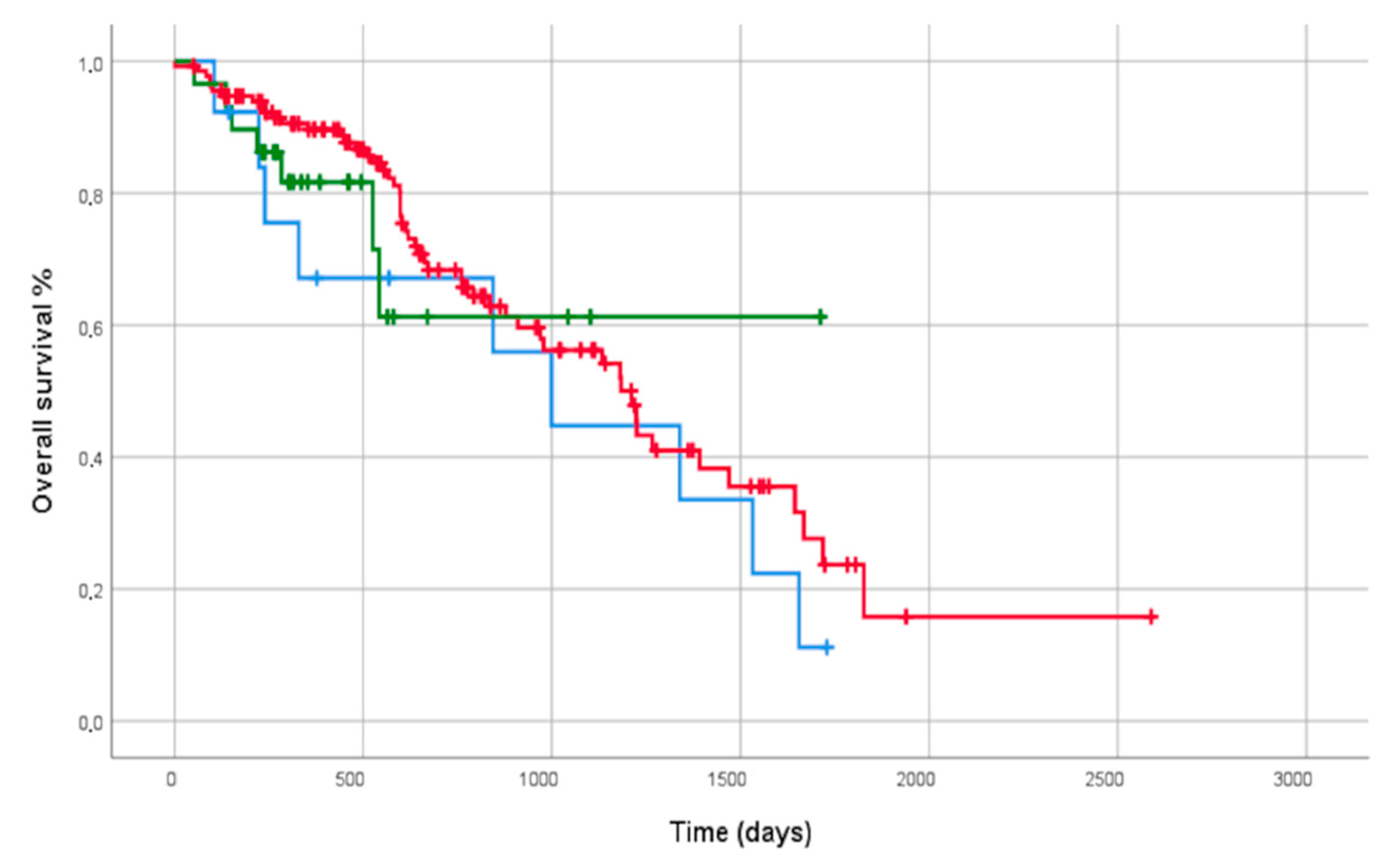
3.4. Outcome Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, S.-L.; Kaiser, R. Nintedanib: From Discovery to the Clinic. J. Med. Chem. 2015, 58, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS® trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Dobashi, M.; Tanaka, H.; Taima, K.; Itoga, M.; Ishioka, Y.; Shiratori, T.; Okumura, F.; Tabe, C.; Tanaka, Y.; Morimoto, T.; et al. The efficacy of nintedanib in 158 patients with idiopathic pulmonary fibrosis in real-world settings: A multicenter retrospective study. SAGE Open Med. 2021, 9, 20503121211023356. [Google Scholar] [CrossRef]
- Rahaghi, F.; Belperio, J.A.; Fitzgerald, J.; Gulati, M.; Hallowell, R.; Highland, K.B.; Huie, T.J.; Kim, H.J.; Kolb, M.; Lasky, J.A.; et al. Delphi Consensus Recommendations on Management of Dosing, Adverse Events, and Comorbidities in the Treatment of Idiopathic Pulmonary Fibrosis with Nintedanib. Clin. Med. Insights Circ. Respir. Pulm. Med. 2021, 15, 11795484211006050. [Google Scholar] [CrossRef]
- Kato, M.; Sasaki, S.; Tateyama, M.; Arai, Y.; Motomura, H.; Sumiyoshi, I.; Ochi, Y.; Watanabe, J.; Ihara, H.; Togo, S.; et al. Clinical Significance of Continuable Treatment with Nintedanib Over 12 Months for Idiopathic Pulmonary Fibrosis in a Real-World Setting. Drug Des. Dev. Ther. 2021, 15, 223–230. [Google Scholar] [CrossRef]
- Antoniou, K.; Markopoulou, K.; Tzouvelekis, A.; Trachalaki, A.; Vasarmidi, E.; Organtzis, J.; Tzilas, V.; Bouros, E.; Kounti, G.; Rampiadou, C.; et al. Efficacy and safety of nintedanib in a Greek multicentre idiopathic pulmonary fibrosis registry: A retrospective, observational, cohort study. ERJ Open Res. 2020, 6, 172–2019. [Google Scholar] [CrossRef]
- Brunnemer, E.; Wälscher, J.; Tenenbaum, S.; Hausmanns, J.; Schulze, K.; Seiter, M.; Heussel, C.P.; Warth, A.; Herth, F.J.F.; Kreuter, M. Real-World Experience with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Respir. Int. Rev. Thorac. Dis. 2018, 95, 301–309. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Karampitsakos, T.; Kontou, M.; Granitsas, A.; Malliou, I.; Anagnostopoulos, A.; Ntolios, P.; Tzilas, V.; Bouros, E.; Steiropoulos, P.; et al. Safety and efficacy of nintedanib in idiopathic pulmonary fibrosis: A real-life observational study in Greece. Pulm. Pharmacol. Ther. 2018, 49, 61–66. [Google Scholar] [CrossRef]
- Hughes, G.; Toellner, H.; Morris, H.; Leonard, C.; Chaudhuri, N. Real World Experiences: Pirfenidone and Nintedanib are Effective and Well Tolerated Treatments for Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2016, 5, 78. [Google Scholar] [CrossRef]
- Sgalla, G.; Greco, E.L.; Calvello, M.; Varone, F.; Iovene, B.; Cerri, S.; Donatelli, P.; Vancheri, A.; Pavone, M.; Luppi, F.; et al. Disease progression across the spectrum of idiopathic pulmonary fibrosis: A multicentre study. Respirol. Carlton Vic. 2020, 25, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Crestani, B.; Hernandez, P.; Inoue, Y.; Wachtlin, D.; Loaiza, L.; Quaresma, M.; Stowasser, S.; Richeldi, L. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: Pooled data from six clinical trials. BMJ Open Respir. Res. 2019, 6, e000397. [Google Scholar] [CrossRef]
- Lawson, W.E.; Loyd, J.E. The genetic approach in pulmonary fibrosis: Can it provide clues to this complex disease? Proc. Am. Thorac. Soc. 2006, 3, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Vold, A.M.; Maher, T.M.; Philpot, E.E.; Ashrafzadeh, A.; Barake, R.; Barsotti, S.; Bruni, C.; Carducci, P.; Carreira, P.E.; Castellví, I.; et al. The identification and management of interstitial lung disease in systemic sclerosis: Evidence-based European consensus statements. Lancet Rheumatol. 2020, 2, e71–e83. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Brown, K.K.; Wells, A.U.; Clerisme-Beaty, E.; Collard, H.R.; Cottin, V.; Devaraj, A.; Inoue, Y.; Le Maulf, F.; Richeldi, L.; et al. Design of the PF-ILD trial: A double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir. Res. 2017, 4, e000212. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.L.; Steenbruggen, I.; Miller, M.R.; Barjaktarevic, I.Z.; Cooper, B.G.; Hall, G.L.; Hallstrand, T.S.; Kaminsky, D.A.; McCarthy, K.; McCormack, M.C.; et al. Standardization of Spirometry 2019 Update. An Official American Thoracic Society and European Respiratory Society Technical Statement. Am. J. Respir. Crit. Care Med. 2019, 200, e70–e88. [Google Scholar] [CrossRef]
- Graham, B.L.; Brusasco, V.; Burgos, F.; Cooper, B.G.; Jensen, R.; Kendrick, A.; MacIntyre, N.R.; Thompson, B.R.; Wanger, J. 2017 ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur. Respir. J. 2017, 49, 1600016. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Refini, R.M.; Bergantini, L.; d’Alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-Term Follow-Up of Patients With Idiopathic Pulmonary Fibrosis Treated With Pirfenidone or Nintedanib: A Real-Life Comparison Study. Front. Mol. Biosci. 2020, 7, 581828. [Google Scholar] [CrossRef] [PubMed]
- Marijic, P.; Schwarzkopf, L.; Schwettmann, L.; Ruhnke, T.; Trudzinski, F.; Kreuter, M. Pirfenidone vs. nintedanib in patients with idiopathic pulmonary fibrosis: A retrospective cohort study. Respir. Res. 2021, 22, 268. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, E.; Piccioli, C.; Rosi, E.; Torricelli, E.; Turi, L.; Piccioli, E.; Ferrari, K.; Voltolini, L. Pirfenidone and Nintedanib in idiopathic pulmonary fibrosis: Real-life experience in an Italian referral centre. Pulmonology 2019, 25, 149–153. [Google Scholar] [CrossRef]
- Belhassen, M.; Dalon, F.; Nolin, M.; Van Ganse, E. Comparative outcomes in patients receiving pirfenidone or nintedanib for idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 135. [Google Scholar] [CrossRef] [PubMed]
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Lic, M.Q.; Stowasser, S.; et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68. [Google Scholar] [CrossRef]
- Song, J.W.; Ogura, T.; Inoue, Y.; Xu, Z.; Quaresma, M.; Stowasser, S.; Stansen, W.; Crestani, B. Long-term treatment with nintedanib in Asian patients with idiopathic pulmonary fibrosis: Results from INPULSIS®-ON. Respirol. Carlton Vic. 2020, 25, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Kreuter, M.; Selman, M.; Crestani, B.; Kirsten, A.M.; Wuyts, W.A.; Xu, Z.; Bernois, K.; Stowasser, S.; Quaresma, M.; et al. Long-term treatment of patients with idiopathic pulmonary fibrosis with nintedanib: Results from the TOMORROW trial and its open-label extension. Thorax 2018, 73, 581–583. [Google Scholar] [CrossRef]
- Fernández-Fabrellas, E.; Registry, O.B.O.T.S.-I.N.; Molina-Molina, M.; Soriano, J.B.; Portal, J.A.R.; Ancochea, J.; Valenzuela, C.; Xaubet, A. Demographic and clinical profile of idiopathic pulmonary fibrosis patients in Spain: The SEPAR National Registry. Respir. Res. 2019, 20, 127. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Šterclová, M.; Mogulkoc, N.; Lewandowska, K.; Müller, V.; Hájková, M.; Kramer, M.R.; Jovanović, D.; Tekavec-Trkanjec, J.; Studnicka, M.; et al. The European MultiPartner IPF registry (EMPIRE): Validating long-term prognostic factors in idiopathic pulmonary fibrosis. Respir. Res. 2020, 21, 11. [Google Scholar] [CrossRef]
- Cottin, V.; Tomassetti, S.; Valenzuela, C.; Walsh, S.L.F.; Antoniou, K.M.; Bonella, F.; Brown, K.K.; Collard, H.R.; Corte, T.J.; Flaherty, K.R.; et al. Integrating Clinical Probability into the Diagnostic Approach to Idiopathic Pulmonary Fibrosis: An International Working Group Perspective. Am. J. Respir. Crit. Care Med. 2022, 206, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.; Larrieu, S.; Si-Mohamed, S.; Ahmad, K.; Boussel, L.; Brevet, M.; Chalabreysse, L.; Fabre, C.; Marque, S.; Revel, D.; et al. Progressive fibrosing interstitial lung disease: A clinical cohort (the PROGRESS study). Eur. Respir. J. 2021, 57, 2002718. [Google Scholar] [CrossRef] [PubMed]
- Hambly, N.; Farooqi, M.M.; Dvorkin-Gheva, A.; Donohoe, K.; Garlick, K.; Scallan, C.; Chong, S.G.; MacIsaac, S.; Assayag, D.; Johannson, K.A.; et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur. Respir. J. 2022, 10, 2102571. [Google Scholar] [CrossRef] [PubMed]
- Tzilas, V.; Tzouvelekis, A.; Bouros, E.; Karampitsakos, T.; Ntassiou, M.; Avdoula, E.; Trachalaki, A.; Antoniou, K.; Raghu, G.; Bouros, D. Clinical experience with antifibrotics in fibrotic hypersensitivity pneumonitis: A 3-year real-life observational study. ERJ Open Res. 2020, 6, 152–2020. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.L.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Justet, A.; Klay, D.; Porcher, R.; Cottin, V.; Ahmad, K.; Molina, M.M.; Nunes, H.; Reynaud-Gaubert, M.; Naccache, J.M.; Manali, E.; et al. Safety and efficacy of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis and carrying a telomere-related gene mutation. Eur. Respir. J. 2021, 57, 2003198. [Google Scholar] [CrossRef]
- Bennett, D.; Refini, R.M.; Valentini, M.L.; Fui, A.; Fossi, A.; Pieroni, M.; Mazzei, M.A.; Rottoli, P. Pirfenidone Therapy for Familial Pulmonary Fibrosis: A Real-Life Study. Lung 2019, 197, 147–153. [Google Scholar] [CrossRef]
- Koga, Y.; Hachisu, Y.; Tsurumaki, H.; Yatomi, M.; Kaira, K.; Ohta, S.; Ono, J.; Izuhara, K.; Dobashi, K.; Hisada, T. Pirfenidone Improves Familial Idiopathic Pulmonary Fibrosis without Affecting Serum Periostin Levels. Med. Kaunas Lith. 2019, 55, 161. [Google Scholar] [CrossRef] [PubMed]
- Planas-Cerezales, L.; Arias-Salgado, E.G.; Buendia-Roldán, I.; Montes-Worboys, A.; López, C.E.; Vicens-Zygmunt, V.; Hernaiz, P.L.; Sanuy, R.L.; Leiro-Fernández, V.; Vilarnau, E.B.; et al. Predictive factors and prognostic effect of telomere shortening in pulmonary fibrosis. Respirology 2019, 24, 146–153. [Google Scholar] [CrossRef]
- Molina-Molina, M.; Xaubet, A.; Li, X.; Abdul-Hafez, A.; Friderici, K.; Jernigan, K.; Fu, W.; Ding, Q.; Pereda, J.; Serrano-Mollar, A.; et al. Angiotensinogen gene G-6A polymorphism influences idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 2008, 32, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response to N-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Study Population | IPF | PF-ILD | FPF | p-Value (Mean Difference; CI 95%) |
|---|---|---|---|---|---|
| N° | 197 | 150 | 30 | 17 | p = 0.0019 * (6.305; 2.152–10.46) |
| Age (yrs) | 72.6 ± 8.1 | 74.4 ± 8.2 | 68.1 ± 9.6 | 70.3 ± 7.7 | p = 0.1579 ° (4.126; −2.117–11.22) |
| Male gender (%) | 145 | 117 (78) | 17 (56.6) | 11 (64.7) | p = 0.0092 * (Chi-square: 9.372) |
| p = 0.0957 ° (Chi-square: 9.372) | |||||
| Pack/year | 28.3 ± 15 | 27.2 ± 14.7 | 30.6 ± 17.3 | 25.1 ±17.5 | p = 0.1869 |
| Comorbidities | |||||
| Hypertension | 81 | 66 | 10 | 5 | p = 0.3283 |
| GERD | 33 | 25 | 5 | 3 | p = 0.9947 |
| Ischemic heart disease | 45 | 33 | 9 | 3 | p = 0.5508 |
| Cancer | 14 | 10 | 3 | 1 | p = 0.8874 |
| Diabetes mellitus | 33 | 22 | 7 | 4 | p = 0.3754 |
| Osteoporosis | 35 | 25 | 6 | 4 | p = 0.7360 |
| HRCT pattern | |||||
| UIP | 121 | 105 | 6 | 10 | p = 0.00004 (Chi-square: 44.77) |
| Probable UIP | 41 | 40 | 1 | 0 | |
| Indeterminate for UIP | 14 | 5 | 4 | 5 | |
| Not UIP | 21 | 0 | 19 | 2 | |
| Baseline PFTs | |||||
| FVC (l) | 2.3 ± 0.7 | 2.4 ± 0.7 | 2.2 ± 0.7 | 2.1 ± 0.9 | p = 0.0657 |
| FVC (%) | 74.2 ± 19.7 | 74.9 ± 18.9 | 71.7 ± 20.6 | 73 ± 22.1 | p = 0.2817 |
| FEV1 (l) | 1.8 ± 0.5 | 1.9 ± 0.5 | 1.8 ± 0.5 | 1.7 ± 0.6 | p = 0.0867 |
| FEV1 (%) | 77.5 ± 20 | 78.6 ± 19 | 75.6 ± 20.8 | 75.3 ± 24.1 | p = 0.3258 |
| TLC (l) | 4.3 ± 1.1 | 4.4 ± 1.2 | 4.1 ± 0.8 | 4 ± 1.1 | p = 0.0783 |
| TLC (%) | 71.2 ± 11.1 | 73.6 ± 17.2 | 68.8 ± 12.6 | 69.5 ± 8.5 | p = 0.1597 |
| DLCO (%) | 42.7 ± 17.3 | 41.4 ± 17.9 | 47.6 ± 17.5 | 40.5 ± 15.6 | p = 0.1357 |
| DLCO/AV (%) | 71.2 ± 23.5 | 68.7 ± 22.3 | 72.8 ± 15.6 | 69.7 ± 16.5 | p = 0.3359 |
| ΔFVC ml pre-treatment | (−256) ± 352 | (−270) ± 393 | (−238) ± 320.8 | (−268) ± 224.5 | p = 0.9486 |
| ΔFVC % pre-treatment | (−8.8) ± 11.3 | (−9.1) ± 12 | (−7.7) ± 9.3 | (−12.5) ± 11.5 | p = 0.6589 |
| ΔDLCO % pre-treatment | (−7.5) ± 15.7 | (−7.7) ±15.1 | (−5.6) ± 18.5 | (−8.5) ± 12.5 | p = 0.8853 |
| Covariates | B | SE | p-Value | HR (95% CI) |
|---|---|---|---|---|
| Univariate analysis | ||||
| Age | 0.015 | 0.015 | 0.330 | 1.01 (0.98–1.04) |
| Female gender | −0.935 | 0.334 | 0.005 | 0.39 (0.2–0.75) |
| Time from onset to diagnosis | −0.002 | 0.004 | 0.653 | 0.99 (0.99–1.01) |
| Diagnosis of IPF | −0.176 | 0.409 | 0.665 | 0.84 (0.37–1.87) |
| FVC > 70% | −0.605 | 0.278 | 0.029 | 0.54 (0.31–0.94) |
| DLCO > 50% | −0.669 | 0.341 | 0.045 | 0.51 (0.26–0.99) |
| paO2/FiO2 > 300 | −1.072 | 0.412 | 0.009 | 0.34 (0.15–0.76) |
| SpO2 > 90% during 6MWT | −0.862 | 0.430 | 0.045 | 0.42 (0.18–0.98) |
| UIP pattern at HRCT | 0.452 | 0.526 | 0.032 | 1.3 (0.78–2.65) |
| Multivariate analysis | ||||
| Age | 0.031 | 0.018 | 0.080 | 1.03 (0.99–1.06) |
| Female gender | −0.912 | 0.399 | 0.022 | 0.40 (0.18–0.87) |
| FVC > 70% | −0.024 | 0.008 | 0.003 | 0.76 (0.54–0.88) |
| DLCO > 50% | −0.022 | 0.356 | 0.469 | 0.86 (0.47–1.33) |
| paO2/FiO2 > 300 | −0.259 | 0.459 | 0.150 | 0.79 (0.25–1.45) |
| SpO2 > 90% during 6MWT | −0.056 | 0.422 | 0.568 | 0.88 (0.55–1.45) |
| UIP pattern at HRCT | 0.156 | 0.436 | 0.458 | 1.2 (0.75–1.78) |
| Covariates | B | SE | p-Value | HR (95% CI) |
|---|---|---|---|---|
| Univariate analysis | ||||
| Age | −0.006 | 0.014 | 0.651 | 0.99 (0.96–1.02) |
| Female gender | −0.500 | 0.257 | 0.025 | 0.60 (0.36–1.01) |
| Time from onset to diagnosis | −0.005 | 0.004 | 0.225 | 0.99 (0.98–1.01) |
| Diagnosis of FPF | 0.990 | 0.310 | 0.005 | 2.69 (1.2–6.3) |
| FVC > 70% | −0.392 | 0.242 | 0.030 | 0.67 (0.32–0.97) |
| DLCO > 50% | −0.159 | 0.279 | 0.568 | 0.85 (0.49–1.47) |
| paO2/FiO2 > 300 | −0.719 | 0.455 | 0.114 | 0.48 (0.20–1.18) |
| SpO2 > 90% during 6MWT | −0.178 | 0.364 | 0.625 | 0.83 (0.41–1.70) |
| UIP pattern at HRCT | 0.115 | 0.195 | 0.277 | 1.2 (0.72–1.65) |
| Multivariate analysis | ||||
| Age | −0.005 | 0.017 | 0.778 | 0.99 (0.96–1.02) |
| Female gender | −0.494 | 0.312 | 0.113 | 0.61 (0.33–1.12) |
| FVC > 70% | −0.361 | 0.266 | 0.175 | 0.63 (0.34–1.34) |
| Diagnosis of FPF | 1.234 | 0.588 | 0.036 | 3.43 (1.08–10.87) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cameli, P.; Alonzi, V.; d’Alessandro, M.; Bergantini, L.; Pordon, E.; Guerrieri, M.; Refini, R.M.; Sestini, P.; Bargagli, E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines 2022, 10, 1973. https://doi.org/10.3390/biomedicines10081973
Cameli P, Alonzi V, d’Alessandro M, Bergantini L, Pordon E, Guerrieri M, Refini RM, Sestini P, Bargagli E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines. 2022; 10(8):1973. https://doi.org/10.3390/biomedicines10081973
Chicago/Turabian StyleCameli, Paolo, Valerio Alonzi, Miriana d’Alessandro, Laura Bergantini, Elena Pordon, Marco Guerrieri, Rosa Metella Refini, Piersante Sestini, and Elena Bargagli. 2022. "The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study" Biomedicines 10, no. 8: 1973. https://doi.org/10.3390/biomedicines10081973
APA StyleCameli, P., Alonzi, V., d’Alessandro, M., Bergantini, L., Pordon, E., Guerrieri, M., Refini, R. M., Sestini, P., & Bargagli, E. (2022). The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines, 10(8), 1973. https://doi.org/10.3390/biomedicines10081973