
Depletion of Mdig Changes Proteomic Profiling in Triple Negative Breast Cancer Cells

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Construction of the CRISPR-Cas9 Vector
2.3. Transfection and Colonies Selection
2.4. Western Blotting
2.5. Preparation of Samples for Mass Spectrometry
2.6. Mass Spectrometry Data Analysis
2.7. Statistical Analysis
2.8. Bioinformatics
2.9. Kaplan–Meier Survival Analysis
3. Results
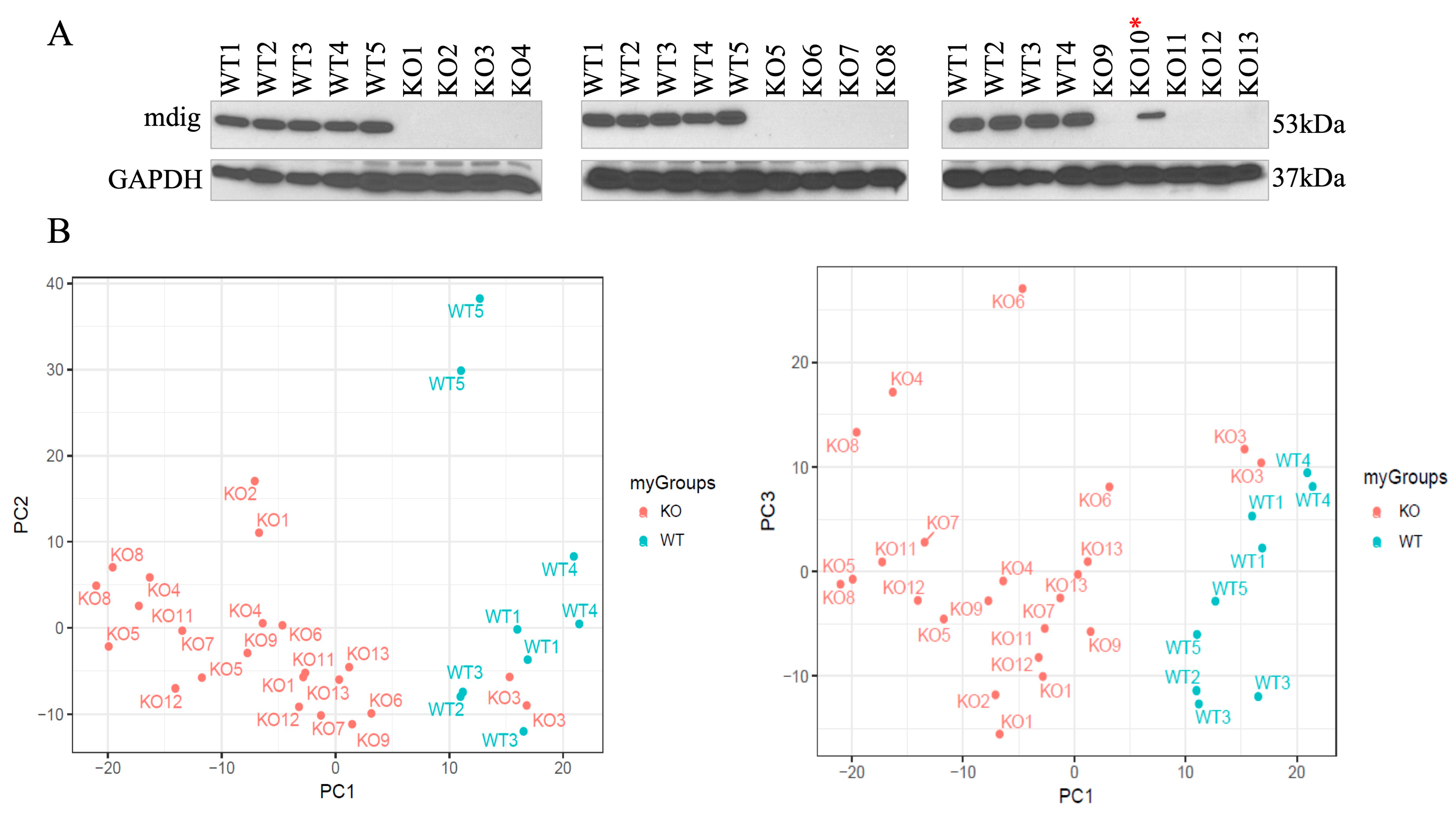
3.1. Generation of Mdig Knockout Cells by CRISPR-Cas9
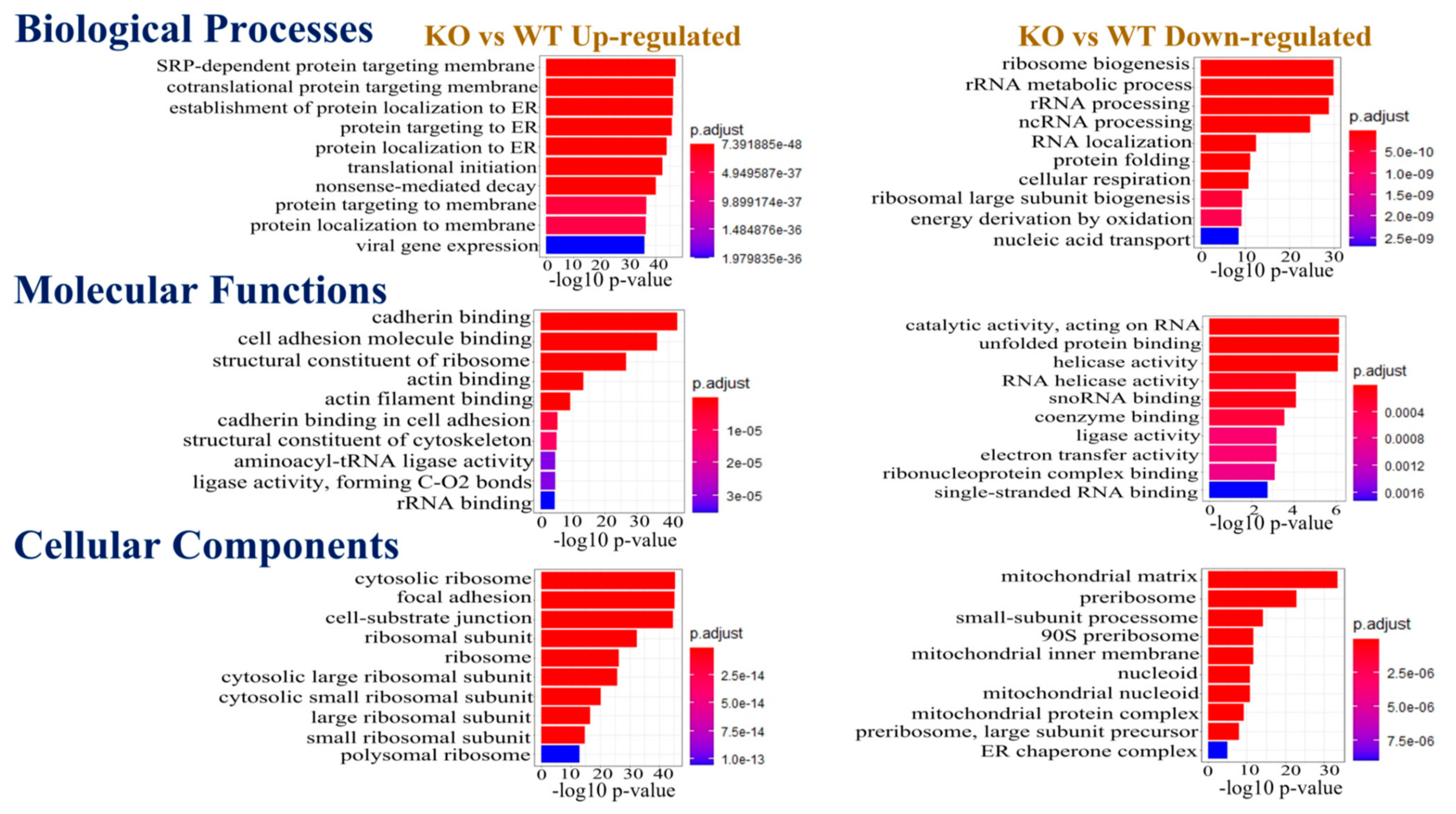
3.2. Identification of the Differentially Expressed Proteins for Their Gene Ontology Annotation
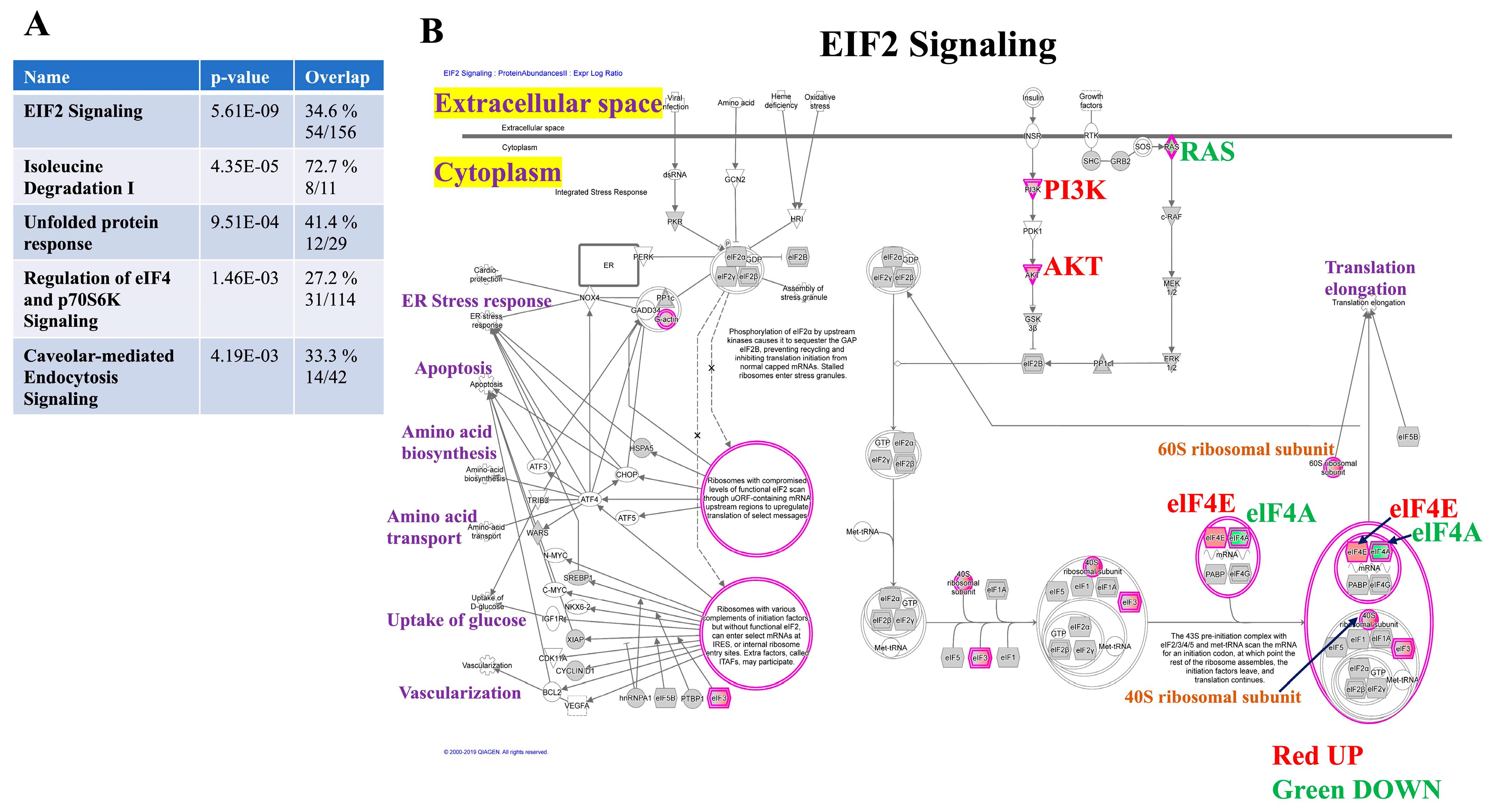
3.3. Canonical Pathway Analysis Reveals Key Signaling Cascades Affected by Mdig
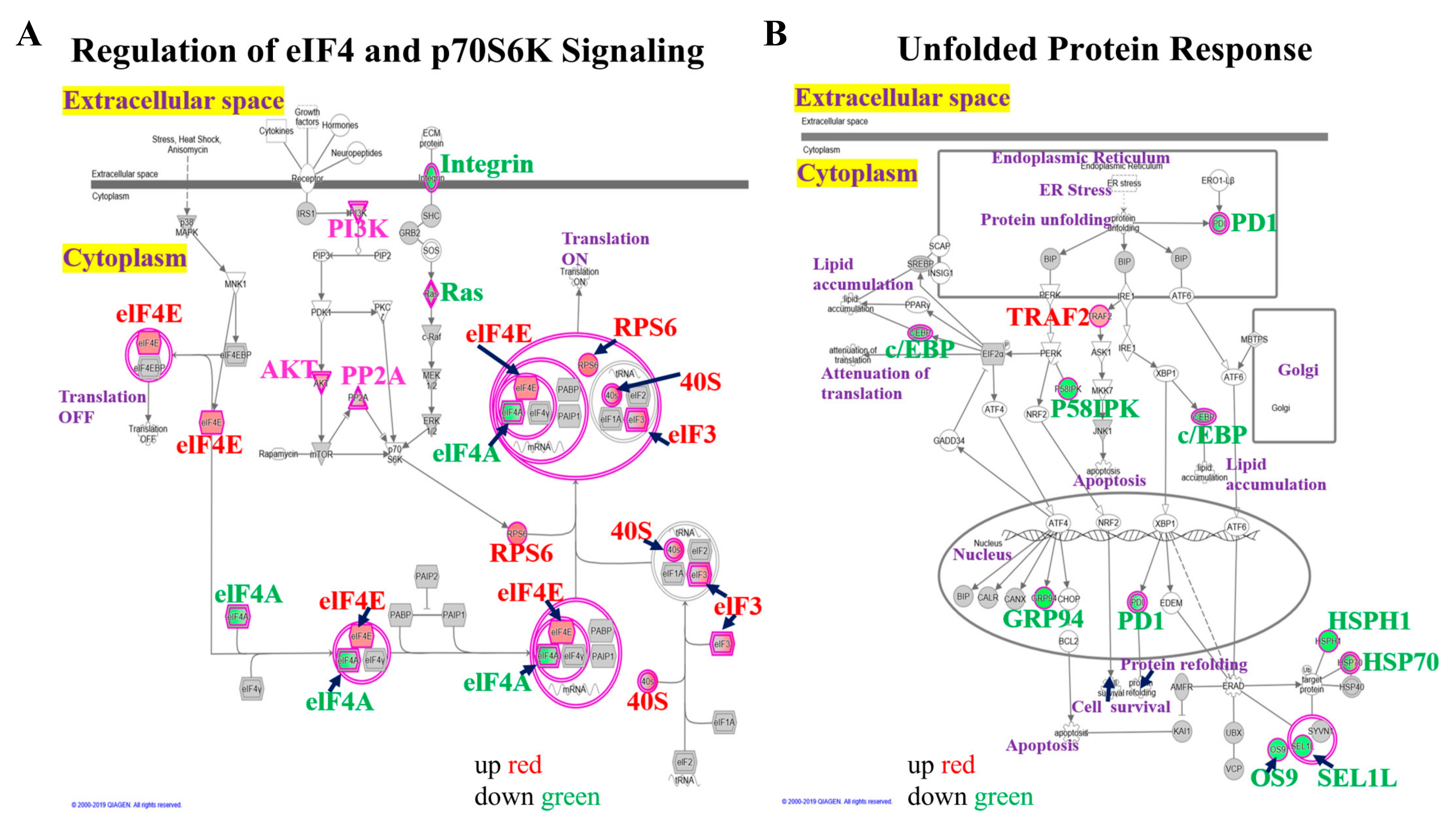
3.4. Upregulation of eIF4-p70S6K and UPR Pathways in Mdig KO Cells
3.5. Disease Pathways Regulated by Mdig
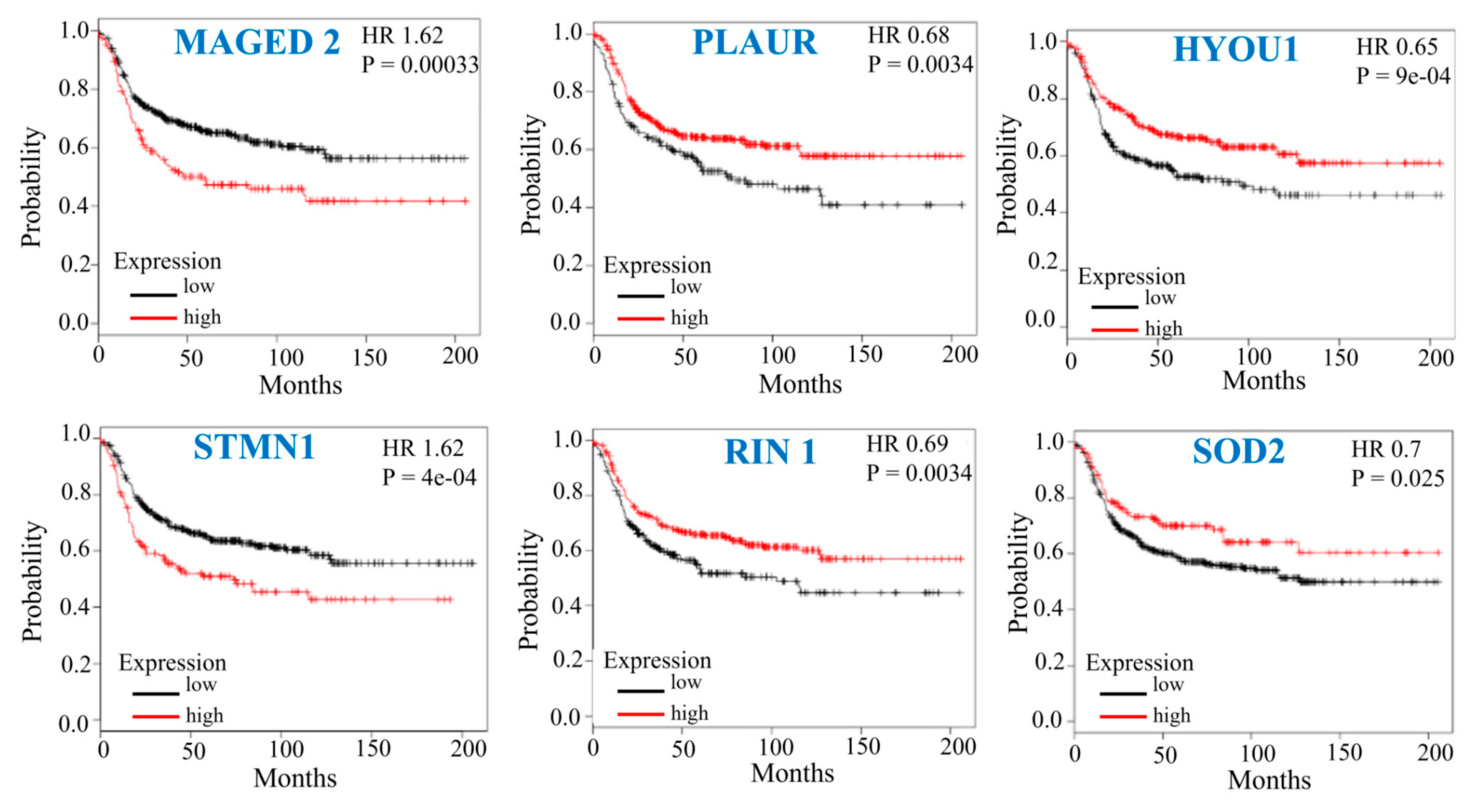
3.6. Prognostic Implication of the Top Mdig-Regulated Genes in Breast Cancer
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.C.; Patek, A.J. The relationship between prognostic and predictive factors in the management of breast cancer. Breast Cancer Res. Treat. 1998, 52, 261–288. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.; Appelgren, J.; Solterbeck, A.; Bergenheim, M.; Alvariza, V.; Bergh, J. Breast cancer during follow-up and progression—A population based cohort on new cancers and changed biology. Eur. J. Cancer 2014, 50, 2916–2924. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Hanna, W.M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Narod, S.A. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res. Treat. 2009, 115, 423–428. [Google Scholar] [CrossRef]
- Madigan, M.P.; Ziegler, R.G.; Benichou, J.; Byrne, C.; Hoover, R.N. Proportion of breast cancer cases in the United States explained by well-established risk factors. J. Natl. Cancer Inst. 1995, 87, 1681–1685. [Google Scholar] [CrossRef]
- Steiner, E.; Klubert, D.; Knutson, D. Assessing breast cancer risk in women. Am. Fam. Physician 2008, 78, 1361–1366. [Google Scholar]
- Yager, J.D.; Davidson, N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef]
- Palmer, J.R.; Rosenberg, L. Cigarette smoking and the risk of breast cancer. Epidemiol. Rev. 1993, 15, 145–156. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Easton, D.F. Models of genetic susceptibility to breast cancer. Oncogene 2006, 25, 5898–5905. [Google Scholar] [CrossRef]
- Greenwald, P. Role of dietary fat in the causation of breast cancer: Point. Cancer Epidemiol Biomarkers Prev. 1999, 8, 3–7. [Google Scholar] [PubMed]
- Thakur, C.; Lu, Y.; Sun, J.; Yu, M.; Chen, B.; Chen, F. Increased expression of mdig predicts poorer survival of the breast cancer patients. Gene 2014, 535, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, B.; Li, L.; Zhang, Q.; Yang, Z.Q.; Chen, F. Loss of mdig expression enhances DNA and histone methylation and metastasis of aggressive breast cancer. Signal Transduct. Target. Ther. 2018, 3, 25. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Y.; Yuan, B.Z.; Castranova, V.; Shi, X.; Stauffer, J.L.; Demers, L.M.; Chen, F. The Human mineral dust-induced gene, mdig, is a cell growth regulating gene associated with lung cancer. Oncogene 2005, 24, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Thakur, C.; Chen, F. Current understanding of mdig/MINA in human cancers. Genes Cancer 2015, 6, 288–302. [Google Scholar] [CrossRef]
- Mori, T.; Okamoto, K.; Tanaka, Y.; Teye, K.; Umata, T.; Ohneda, K.; Tokuyama, K.; Okabe, M.; Tsuneoka, M. Ablation of Mina53 in mice reduces allergic response in the airways. Cell Struct. Funct. 2013, 38, 155–167. [Google Scholar] [CrossRef]
- Thakur, C.; Wolfarth, M.; Sun, J.; Zhang, Y.; Lu, Y.; Battelli, L.; Porter, D.W.; Chen, F. Oncoprotein mdig contributes to silica-induced pulmonary fibrosis by altering balance between Th17 and Treg T cells. Oncotarget 2015, 6, 3722–3736. [Google Scholar] [CrossRef]
- Hemmers, S.; Mowen, K.A. T(H)2 bias: Mina tips the balance. Nat. Immunol. 2009, 10, 806–808. [Google Scholar] [CrossRef]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef]
- Yu, M.; Sun, J.; Thakur, C.; Chen, B.; Lu, Y.; Zhao, H.; Chen, F. Paradoxical roles of mineral dust induced gene on cell proliferation and migration/invasion. PLoS ONE 2014, 9, e87998. [Google Scholar] [CrossRef]
- Sun, J.; Yu, M.; Lu, Y.; Thakur, C.; Chen, B.; Qiu, P.; Zhao, H.; Chen, F. Carcinogenic metalloid arsenic induces expression of mdig oncogene through JNK and STAT3 activation. Cancer Lett. 2014, 346, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Thakur, C.; Fu, Y.; Bi, Z.; Wadgaonkar, P.; Xu, L.; Liu, Z.; Liu, W.; Wang, J.; Kidder, B.L.; et al. Mdig promotes oncogenic gene expression through antagonizing repressive histone methylation markers. Theranostics 2020, 10, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.D.; Fang, Y.; Foster, L.J. An integrated global strategy for cell lysis, fractionation, enrichment and mass spectrometric analysis of phosphorylated peptides. Mol. Biosyst. 2010, 6, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article3. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Li, Q.; Birkbak, N.J.; Gyorffy, B.; Szallasi, Z.; Eklund, A.C. Jetset: Selecting the optimal microarray probe set to represent a gene. BMC Bioinform. 2011, 12, 474. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Qiu, Y.; Shabason, J.E.; Wu, T.J.; Yoon, T.; Kim, B.C.; Benci, J.L.; DeMichele, A.M.; Tchou, J.; Marcotrigiano, J.; et al. Exosome RNA Unshielding Couples Stromal Activation to Pattern Recognition Receptor Signaling in Cancer. Cell 2017, 170, 352–366.e313. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, M. Gene Dosage Analysis on the Single-Cell Transcriptomes Linking Cotranslational Protein Targeting to Metastatic Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 918. [Google Scholar] [CrossRef]
- Verma, M.; Choi, J.; Cottrell, K.A.; Lavagnino, Z.; Thomas, E.N.; Pavlovic-Djuranovic, S.; Szczesny, P.; Piston, D.W.; Zaher, H.S.; Puglisi, J.D.; et al. A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 2019, 10, 5774. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014, 83, 779–812. [Google Scholar] [CrossRef]
- Ali, M.U.; Ur Rahman, M.S.; Jia, Z.; Jiang, C. Eukaryotic translation initiation factors and cancer. Tumor. Biol. 2017, 39, 1010428317709805. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.; Borden, K.L. The oncogene eIF4E: Using biochemical insights to target cancer. J. Interferon Cytokine Res. 2013, 33, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, P.B.; Polunovsky, V.A. eIF4E-mediated translational control of cancer incidence. Biochim. Biophys. Acta 2015, 1849, 774–780. [Google Scholar] [CrossRef]
- Heikkinen, T.; Korpela, T.; Fagerholm, R.; Khan, S.; Aittomäki, K.; Heikkilä, P.; Blomqvist, C.; Carpén, O.; Nevanlinna, H. Eukaryotic translation initiation factor 4E (eIF4E) expression is associated with breast cancer tumor phenotype and predicts survival after anthracycline chemotherapy treatment. Breast Cancer Res. Treat. 2013, 141, 79–88. [Google Scholar] [CrossRef]
- Nasr, Z.; Robert, F.; Porco, J.A., Jr.; Muller, W.J.; Pelletier, J. eIF4F suppression in breast cancer affects maintenance and progression. Oncogene 2013, 32, 861–871. [Google Scholar] [CrossRef]
- Flowers, A.; Chu, Q.D.; Panu, L.; Meschonat, C.; Caldito, G.; Lowery-Nordberg, M.; Li, B.D. Eukaryotic initiation factor 4E overexpression in triple-negative breast cancer predicts a worse outcome. Surgery 2009, 146, 220–226. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Mallini, P.; Lennard, T.; Kirby, J.; Meeson, A. Epithelial-to-mesenchymal transition: What is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. 2014, 40, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Zutter, M.M.; Krigman, H.R.; Santoro, S.A. Altered integrin expression in adenocarcinoma of the breast. Analysis by in situ hybridization. Am. J. Pathol. 1993, 142, 1439–1448. [Google Scholar] [PubMed]
- Belin, S.; Beghin, A.; Solano-Gonzàlez, E.; Bezin, L.; Brunet-Manquat, S.; Textoris, J.; Prats, A.C.; Mertani, H.C.; Dumontet, C.; Diaz, J.J. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS ONE 2009, 4, e7147. [Google Scholar] [CrossRef] [PubMed]
- Yanai, A.; Inoue, N.; Yagi, T.; Nishimukai, A.; Miyagawa, Y.; Murase, K.; Imamura, M.; Enomoto, Y.; Takatsuka, Y.; Watanabe, T.; et al. Activation of mTOR/S6K But Not MAPK Pathways Might Be Associated With High Ki-67, ER(+), and HER2(-) Breast Cancer. Clin. Breast Cancer 2015, 15, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int. Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar] [CrossRef]
- Papaioannou, A.; Chevet, E. Driving Cancer Tumorigenesis and Metastasis Through UPR Signaling. Curr. Top Microbiol. Immunol. 2018, 414, 159–192. [Google Scholar] [CrossRef]
- Peramuhendige, P.; Marino, S.; Bishop, R.T.; de Ridder, D.; Khogeer, A.; Baldini, I.; Capulli, M.; Rucci, N.; Idris, A.I. TRAF2 in osteotropic breast cancer cells enhances skeletal tumour growth and promotes osteolysis. Sci. Rep. 2018, 8, 39. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Ren, H.Y.; Yang, F.; Wang, J.; Wu, G.P.; Mi, X.Y. Expression, correlation, and prognostic value of TRAF2 and TRAF4 expression in malignant plural effusion cells in human breast cancer. Diagn. Cytopathol. 2015, 43, 897–903. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Mamessier, E.; Adélaïde, J.; Debono, S.; Houvenaeghel, G.; Maraninchi, D.; Viens, P.; et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006, 66, 4636–4644. [Google Scholar] [CrossRef]
- Nagy, Á.; Lánczky, A.; Menyhárt, O.; Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chang, Q.; Zhang, Y.; Beezhold, K.; Rojanasakul, Y.; Zhao, H.; Castranova, V.; Shi, X.; Chen, F. Lung cancer-associated JmjC domain protein mdig suppresses formation of tri-methyl lysine 9 of histone H3. Cell Cycle 2009, 8, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Platet, N.; Liaudet, E.; Laurent, V.; Derocq, D.; Brouillet, J.-P.; Rochefort, H. Biological and Clinical Significance of Cathepsin D in Breast Cancer Metastasis. Stem Cells 1996, 14, 642–650. [Google Scholar] [CrossRef]
- Capony, F.; Rougeot, C.; Montcourrier, P.; Cavailles, V.; Salazar, G.; Rochefort, H. Increased secretion, altered processing, and glycosylation of pro-cathepsin D in human mammary cancer cells. Cancer Res. 1989, 49, 3904–3909. [Google Scholar]
- Rochefort, H.; Cavailles, V.; Augereau, P.; Capony, F.; Maudelonde, T.; Touitou, I.; Garcia, M. Overexpression and hormonal regulation of pro-cathepsin D in mammary and endometrial cancer. J. Steroid Biochem. 1989, 34, 177–182. [Google Scholar] [CrossRef]
- Rochefort, H. Cathepsin D in breast cancer: A tissue marker associated with metastasis. Eur. J. Cancer 1992, 28, 1780–1783. [Google Scholar] [CrossRef]
- Ferrandina, G.; Scambia, G.; Bardelli, F.; Benedetti Panici, P.; Mancuso, S.; Messori, A. Relationship between cathepsin-D content and disease-free survival in node-negative breast cancer patients: A meta-analysis. Br. J. Cancer 1997, 76, 661–666. [Google Scholar] [CrossRef]
- Westley, B.R.; May, F.E. Prognostic value of cathepsin D in breast cancer. Br. J. Cancer 1999, 79, 189–190. [Google Scholar] [CrossRef]
- Sivaranjini, Y.; Angadi, P.V.; Kumar, G.S. Immuno-histochemical evaluation of Cathepsin D in malignant salivary gland carcinomas. Gulf J. Oncol. 2012, 11, 38–44. [Google Scholar]
- Ohri, S.S.; Vashishta, A.; Proctor, M.; Fusek, M.; Vetvicka, V. The propeptide of cathepsin D increases proliferation, invasion and metastasis of breast cancer cells. Int. J. Oncol. 2008, 32, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Vashishta, A.; Ohri, S.S.; Proctor, M.; Fusek, M.; Vetvicka, V. Ribozyme-targeting procathepsin D and its effect on invasion and growth of breast cancer cells: An implication in breast cancer therapy. Int. J. Oncol. 2007, 30, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Berchem, G.; Glondu, M.; Gleizes, M.; Brouillet, J.P.; Vignon, F.; Garcia, M.; Liaudet-Coopman, E. Cathepsin-D affects multiple tumor progression steps in vivo: Proliferation, angiogenesis and apoptosis. Oncogene 2002, 21, 5951–5955. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Modlin, I.M.; Mane, S.M.; Camp, R.L.; Eick, G.; Latich, I. The role of genetic markers—NAP1L1, MAGE-D2, and MTA1--in defining small-intestinal carcinoid neoplasia. Ann. Surg. Oncol. 2006, 13, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Curmi, P.A.; Nogues, C.; Lachkar, S.; Carelle, N.; Gonthier, M.P.; Sobel, A.; Lidereau, R.; Bieche, I. Overexpression of stathmin in breast carcinomas points out to highly proliferative tumours. Br. J. Cancer 2000, 82, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Drury, S.; Afentakis, M.; A’Hern, R.; Hills, M.; Salter, J.; Smith, I.E.; Reis-Filho, J.S.; Dowsett, M. Biomarker changes associated with the development of resistance to aromatase inhibitors (AIs) in estrogen receptor-positive breast cancer. Ann. Oncol. 2014, 25, 605–610. [Google Scholar] [CrossRef]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmstrom, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef]
- Baquero, M.T.; Hanna, J.A.; Neumeister, V.; Cheng, H.; Molinaro, A.M.; Harris, L.N.; Rimm, D.L. Stathmin expression and its relationship to microtubule-associated protein tau and outcome in breast cancer. Cancer 2012, 118, 4660–4669. [Google Scholar] [CrossRef]
- Obayashi, S.; Horiguchi, J.; Higuchi, T.; Katayama, A.; Handa, T.; Altan, B.; Bai, T.; Bao, P.; Bao, H.; Yokobori, T.; et al. Stathmin1 expression is associated with aggressive phenotypes and cancer stem cell marker expression in breast cancer patients. Int. J. Oncol. 2017, 51, 781–790. [Google Scholar] [CrossRef]
- Cao, X.-X.; Xu, J.-D.; Xu, J.-W.; Liu, X.-L.; Cheng, Y.-Y.; Wang, W.-J.; Li, Q.-Q.; Chen, Q.; Xu, Z.-D.; Liu, X.-P. RACK1 promotes breast carcinoma proliferation and invasion/metastasis in vitro and in vivo. Breast Cancer Res. Treat. 2010, 123, 375–386. [Google Scholar] [CrossRef]
- Cao, X.X.; Xu, J.D.; Liu, X.L.; Xu, J.W.; Wang, W.J.; Li, Q.Q.; Chen, Q.; Xu, Z.D.; Liu, X.P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int. J. Cancer 2010, 127, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.-L.; Huang, Y.-T.; Wang, G.-H.; Liu, Y.-L.; Huang, J.; Qu, Q.; Sun, B.; Hu, L.; Cheng, L.; Chen, S.-H.; et al. Overexpression of RACK1 Promotes Metastasis by Enhancing Epithelial-Mesenchymal Transition and Predicts Poor Prognosis in Human Glioma. Int. J. Environ. Res. Public Health 2016, 13, 1021. [Google Scholar] [CrossRef] [PubMed]
- Bedolla, R.; Wang, Y.; Asuncion, A.; Chamie, K.; Siddiqui, S.; Mudryj, M.; Prihoda, T.; Siddiqui, J.; Chinnaiyan, A.; Mehra, R. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: Immunohistochemical correlation with metastases. Clin. Cancer Res. 2009, 15, 788–796. [Google Scholar] [CrossRef]
- Coughlin, M.; Puig-de-Morales, M.; Bursac, P.; Mellema, M.; Millet, E.; Fredberg, J. Filamin-A and rheological properties of cultured melanoma cells. Biophys. J. 2006, 90, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.; Howard, J.; Vogel, C. Actin-binding protein filamin A is displayed on the surface of human neuroblastoma cells. Cancer Sci. 2006, 97, 1359–1365. [Google Scholar] [CrossRef]
- Yue, J.; Huhn, S.; Shen, Z. Complex roles of filamin-A mediated cytoskeleton network in cancer progression. Cell Biosci. 2013, 3, 7. [Google Scholar] [CrossRef]
- Sharma, D.K.; Bressler, K.; Patel, H.; Balasingam, N.; Thakor, N. Role of Eukaryotic Initiation Factors during Cellular Stress and Cancer Progression. J. Nucleic Acids 2016, 2016, 19. [Google Scholar] [CrossRef]
- Gelder, M.E.M.-v.; Look, M.P.; Peters, H.A.; Schmitt, M.; Brünner, N.; Harbeck, N.; Klijn, J.G.M.; Foekens, J.A. Urokinase-Type Plasminogen Activator System in Breast Cancer. Assoc. Tamoxifen Ther. Recurr. Dis. 2004, 64, 4563–4568. [Google Scholar] [CrossRef]
- Bianchi, E.; Cohen, R.L.; Thor, A.T.; Todd, R.F.; Mizukami, I.F.; Lawrence, D.A.; Ljung, B.M.; Shuman, M.A.; Smith, H.S. The Urokinase Receptor Is Expressed in Invasive Breast Cancer but not in Normal Breast Tissue. Cancer Res. 1994, 54, 861–866. [Google Scholar]
- Konecny, G.; Untch, M.; Arboleda, J.; Wilson, C.; Kahlert, S.; Boettcher, B.; Felber, M.; Beryt, M.; Lude, S.; Hepp, H.; et al. HER-2/neu and Urokinase-Type Plasminogen Activator and Its Inhibitor in Breast Cancer. Clin. Cancer Res. 2001, 7, 2448–2457. [Google Scholar]
- Kim, Y.S.; Gupta Vallur, P.; Phaeton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Oberley, L.W.; St Clair, D.K.; Ridnour, L.A.; Oberley, T.D. Phenotypic changes induced in human breast cancer cells by overexpression of manganese-containing superoxide dismutase. Oncogene 1995, 10, 1989–2000. [Google Scholar] [PubMed]
- Weydert, C.; Roling, B.; Liu, J.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol. Cancer Ther. 2003, 2, 361–369. [Google Scholar] [PubMed]
- Hitchler, M.J.; Oberley, L.W.; Domann, F.E. Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells. Free Radic. Biol. Med. 2008, 45, 1573–1580. [Google Scholar] [CrossRef]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 switch in breast cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.; Wikainapakul, K.; Yu, L.; Powers, K.; Attatippaholkun, W.; Domann, F. Epigenetic Regulation of Manganese Superoxide Dismutase Expression in Human Breast Cancer Cells. Epigenetics 2006, 1, 163–171. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Protein ID | Description | Fold Change |
|---|---|---|---|
| CTSD | P07339 | Cathepsin D | 11.063 |
| MAGED2 | Q9UNF1 | Melanoma-associated antigen D2 | 10.443 |
| FLNA | P21333 | Filamin-A | 9.662 |
| ABHD16A | O95870 | Abhydrolase domain-containing protein 16A | 9.595 |
| STMN1 | P16949 | Stathmin1 | 9.118 |
| NPC2 | P61916 | Epididymal secretory protein E1 | 8.845 |
| RACK1 | P63244 | Receptor of activated protein C kinase 1 | 8.820 |
| HIST1H2BA | Q96A08 | Histone H2B type 1-A | 8.673 |
| IQGAP1 | P46940 | Ras GTPase-activating-like protein IQGAP1 | 8.603 |
| HUWE1 | Q7Z6Z7 | E3 ubiquitin-protein ligase HUWE1 | 8.473 |
| Mdig (RIOX2) | Q8IUF8 | Mineral dust-induced gene | −8.521 |
| KRI1 | Q8N9T8 | Protein KRI1 homolog | −8.272 |
| CCDC51 | Q96ER9 | Coiled-coil domain-containing protein 51 | −8.241 |
| PLAUR | Q03405 | Urokinase plasminogen activator surface receptor | −8.203 |
| HYOU1 | Q9Y4L1 | Hypoxia upregulated protein 1 | −7.368 |
| SOD2 | P04179 | Superoxide dismutase [Mn], mitochondrial | −7.173 |
| RIN1 | Q13671 | Ras and Rab interactor 1 | −7.096 |
| NOP58 | Q9Y2X3 | Nucleolar protein 58 | −7.083 |
| NAMPT | P43490 | Nicotinamide phosphoribosyltransferase | −7.006 |
| MCCC2 | Q9HCC0 | Methylcrotonoyl-CoA carboxylase beta chain, mitochondrial | −6.863 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thakur, C.; Carruthers, N.J.; Zhang, Q.; Xu, L.; Fu, Y.; Bi, Z.; Qiu, Y.; Zhang, W.; Wadgaonkar, P.; Almutairy, B.; et al. Depletion of Mdig Changes Proteomic Profiling in Triple Negative Breast Cancer Cells. Biomedicines 2022, 10, 2021. https://doi.org/10.3390/biomedicines10082021
Thakur C, Carruthers NJ, Zhang Q, Xu L, Fu Y, Bi Z, Qiu Y, Zhang W, Wadgaonkar P, Almutairy B, et al. Depletion of Mdig Changes Proteomic Profiling in Triple Negative Breast Cancer Cells. Biomedicines. 2022; 10(8):2021. https://doi.org/10.3390/biomedicines10082021
Chicago/Turabian StyleThakur, Chitra, Nicholas J. Carruthers, Qian Zhang, Liping Xu, Yao Fu, Zhuoyue Bi, Yiran Qiu, Wenxuan Zhang, Priya Wadgaonkar, Bandar Almutairy, and et al. 2022. "Depletion of Mdig Changes Proteomic Profiling in Triple Negative Breast Cancer Cells" Biomedicines 10, no. 8: 2021. https://doi.org/10.3390/biomedicines10082021