

Eltanexor Effectively Reduces Viability of Glioblastoma and Glioblastoma Stem-Like Cells at Nano-Molar Concentrations and Sensitizes to Radiotherapy and Temozolomide


 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Viability Assay
2.4. Apoptosis Assay
2.5. FACS Analysis
2.6. Radiation Exposure
2.7. Colony Formation Assay
2.8. Immunofluorescence Staining
2.9. RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
2.10. Protein Extraction and Western Blot Analysis
2.11. Migration Assay
2.12. Data Analysis
3. Results
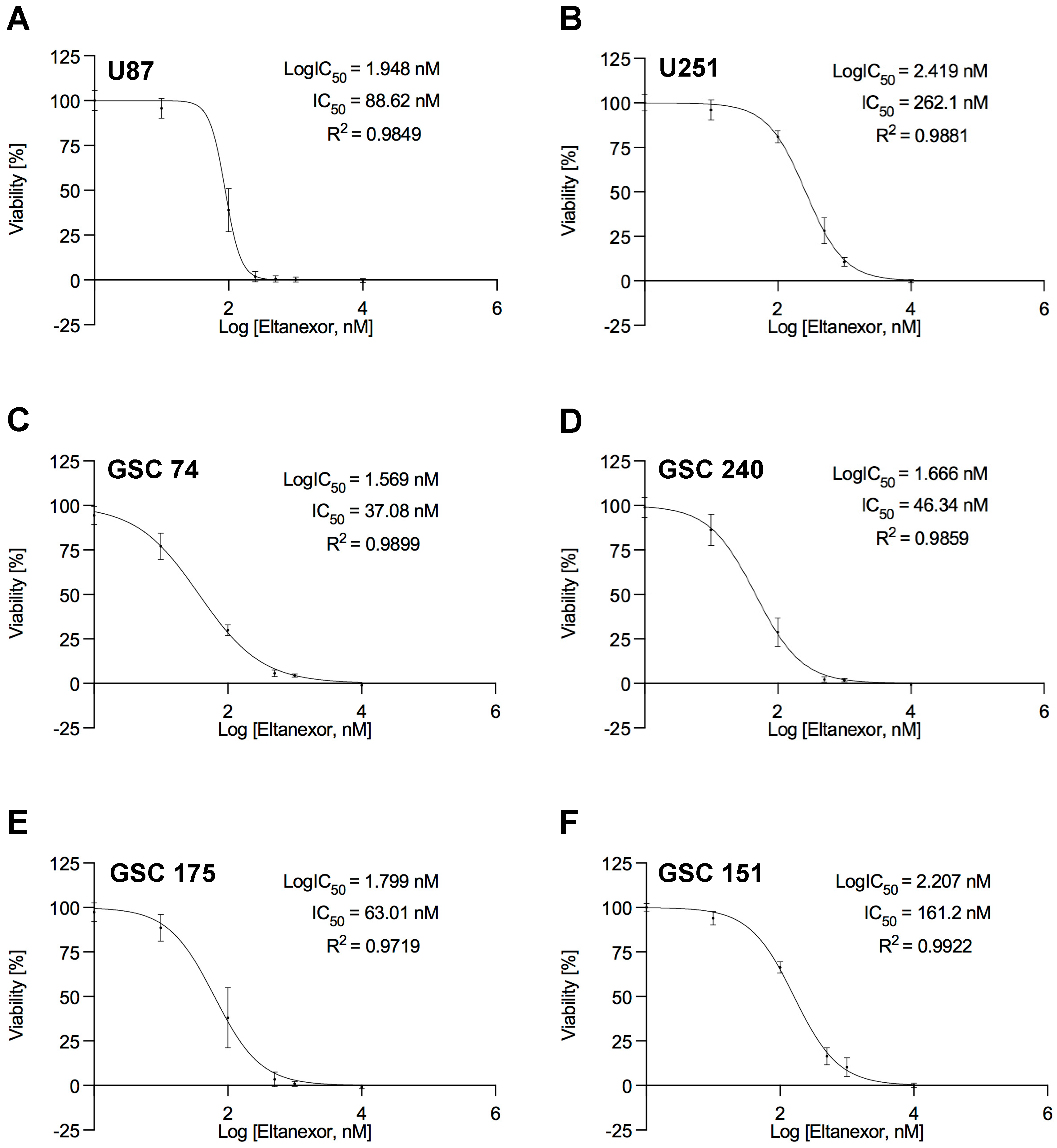
3.1. Reduced Cell Viability of GBM Cells after Treatment with the XPO1 Inhibitor Eltanexor
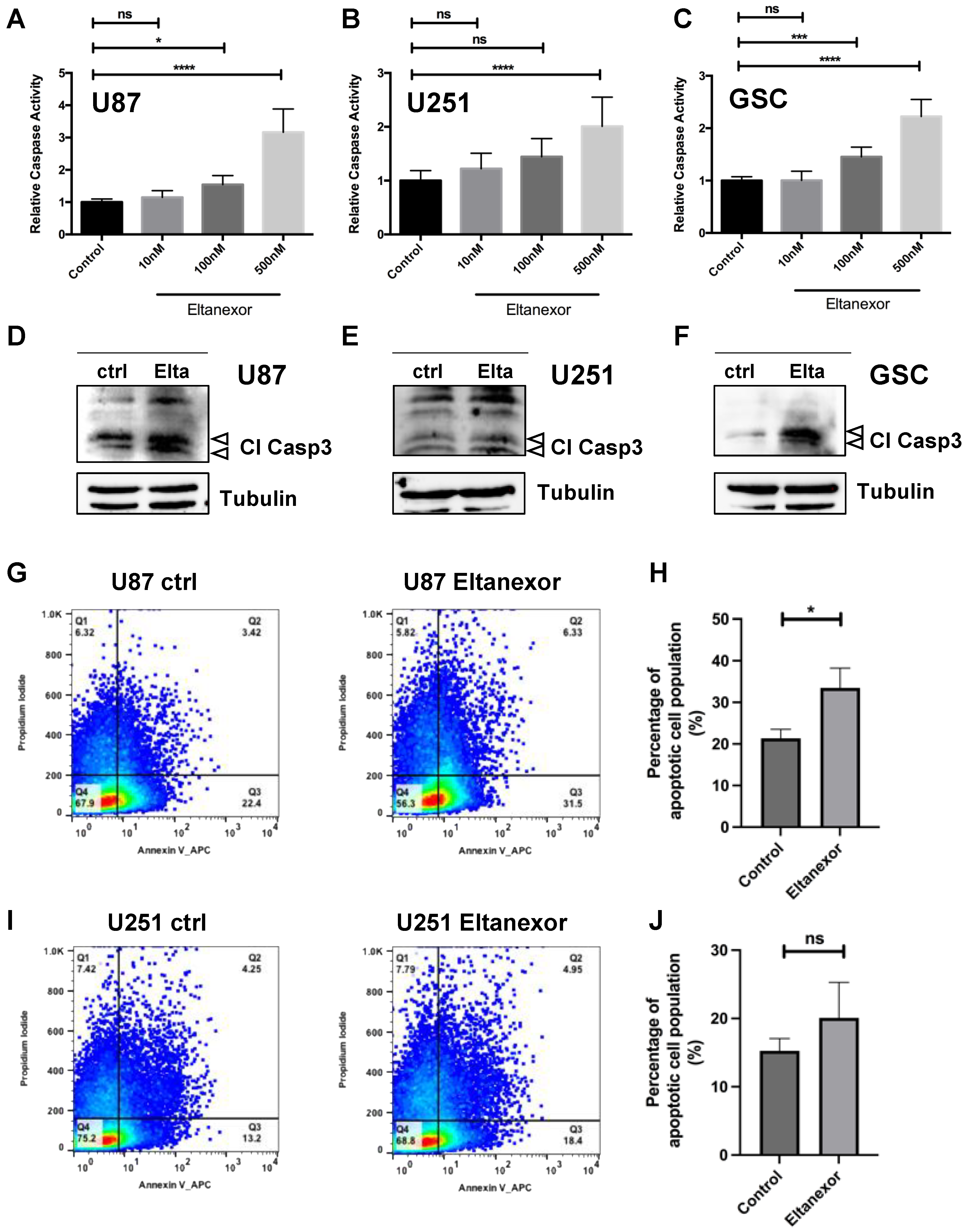
3.2. Eltanexor Induces Apoptosis in GBM Cells by Increased TP53 Signaling
3.3. Eltanexor Causes p53 and CDKN1A to Be Retained in the Nucleus of GBM Cells
3.4. Co-Treatment of Eltanexor with Temozolomide (TMZ)
3.5. Eltanexor Sensitizes GBM Cells to Radiotherapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncology 2013, 15 (Suppl. S2), ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Dehler, S.; Rushing, E.J.; Zaugg, K.; Hofer, S.; Yonekawa, Y.; Bertalanffy, H.; Valavanis, A.; Korol, D.; Rohrmann, S.; et al. Glioblastoma in the Canton of Zurich, Switzerland revisited: 2005 to 2009. Cancer 2016, 122, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Rajaratnam, V.; Islam, M.M.; Yang, M.; Slaby, R.; Ramirez, H.M.; Mirza, S.P. Glioblastoma: Pathogenesis and current status of chemotherapy and other novel treatments. Cancers 2020, 12, 937. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Taphoorn, M.J.; Sizoo, E.M.; Bottomley, A. Review on quality of life issues in patients with primary brain tumors. Oncologist 2010, 15, 618–626. [Google Scholar] [CrossRef]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Burster, T.; Traut, R.; Yermekkyzy, Z.; Mayer, K.; Westhoff, M.A.; Bischof, J.; Knippschild, U. Critical View of Novel Treatment Strategies for Glioblastoma: Failure and Success of Resistance Mechanisms by Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 695325. [Google Scholar] [CrossRef]
- Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6385. [Google Scholar] [CrossRef]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Mollapour Sisakht, M.; Azizi, Z.; Taghikhani, A.; Aos, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updates 2019, 42, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Schäfer, A.; Zhang, Z.; Elsässer, K.; Culmsee, C.; Zhong, L.; Pagenstecher, A.; Nimsky, C.; Bartsch, J.W. Inhibition of Carbonic Anhydrase 2 Overcomes Temozolomide Resistance in Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23, 157. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The controversial role of microglia in malignant gliomas. Clin. Dev. Immunol. 2013, 2013, 285246. [Google Scholar] [CrossRef] [PubMed]
- Roszman, T.L.; Brooks, W.H. Immunobiology of primary intracranial tumours. III. Demonstration of a qualitative lymphocyte abnormality in patients with primary brain tumours. Clin. Exp. Immunol. 1980, 39, 395–402. [Google Scholar] [PubMed]
- Brooks, W.H.; Netsky, M.G.; Normansell, D.E.; Horwitz, D.A. Depressed cell-mediated immunity in patients with primary intracranial tumours. J. Exp. Med. 1972, 136, 1631–1647. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef]
- Perng, P.; Lim, M. Immunosuppressive mechanisms of malignant gliomas: Parallels at Non-CNS sites. Front. Oncol. 2015, 5, 153. [Google Scholar] [CrossRef]
- Fontana, A.; Hengartner, H.; de Tribolet, N.; Weber, E. Glioblastoma cells release interleukin 1 and factors inhibiting interleukin 2-mediated effects. J. Immunol. 1984, 132, 1837–1844. [Google Scholar]
- Siepl, C.; Bodmer, S.; Frei, K.; MacDonald, H.R.; De Martin, R.; Hofer, E.; Fontana, A. The glioblastoma-derived T cell suppressor factor/transforming growth factor-beta 2 inhibits T cell growth without affecting the interaction of interleukin 2 with its receptor. Eur. J. Immunol. 1988, 18, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Kotsarini, C.; Griffiths, P.D.; Wilkinson, I.D.; Hoggard, N. A systematic review of the literature on the effects of dexamethasone on the brain from in vivo human-based studies: Implications for physiological brain imaging of patients with intracranial tumors. Neurosurgery 2010, 67, 1799–1815. [Google Scholar] [CrossRef] [PubMed]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Orzan, F.; De Bacco, F.; Crisafulli, G.; Pellegatta, S.; Mussolin, B.; Siravegna, G.; D’Ambrosio, A.; Comoglio, P.M.; Finocchiaro, G.; Boccaccio, C. Genetic Evolution of Glioblastoma Stem-Like Cells from Primary to Recurrent Tumor. Stem Cells 2017, 35, 2218–2228. [Google Scholar] [CrossRef]
- Stieber, D.; Golebiewska, A.; Evers, L.; Lenkiewicz, E.; Brons, N.H.; Nicot, N.; Oudin, A.; Bougnaud, S.; Hertel, F.; Bjerkvig, R.; et al. Glioblastomas are composed of genetically divergent clones with distinct tumorigenic potential and variable stem cell-associated phenotypes. Acta Neuropathol. 2014, 127, 203–219. [Google Scholar] [CrossRef]
- Dey, M.; Ulasov, I.V.; Lesniak, M.S. Virotherapy against malignant glioma stem cells. Cancer Lett. 2010, 289, 1–10. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; Amiji, M.M. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opin. Drug Deliv. 2017, 14, 997–1008. [Google Scholar] [CrossRef]
- Balcer-Kubiczek, E.K. Apoptosis in radiation therapy: A double-edged sword. Exp. Oncol. 2012, 34, 277–285. [Google Scholar]
- Günther, W.; Pawlak, E.; Damasceno, R.; Arnold, H.; Terzis, A.J. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br. J. Cancer 2003, 88, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; Westhoff, M.A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltzig, L.; Schwarzenbach, C.; Leukel, P.; Frauenknecht, K.B.M.; Sommer, C.; Tancredi, A.; Hegi, M.E.; Christmann, M.; Kaina, B. Senescence Is the Main Trait Induced by Temozolomide in Glioblastoma Cells. Cancers 2022, 14, 2233. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, C.; Tatsch, L.; Brandstetter Vilar, J.; Rasenberger, B.; Beltzig, L.; Kaina, B.; Tomicic, M.T.; Christmann, M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 2021, 13, 3585. [Google Scholar] [CrossRef] [PubMed]
- Hutten, S.; Kehlenbach, R.H. CRM1-mediated nuclear export: To the pore and beyond. Trends Cell Biol. 2007, 17, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 61. [Google Scholar] [CrossRef]
- Wozniak, R.; Burke, B.; Doye, V. Nuclear transport and the mitotic apparatus: An evolving relationship. Cell Mol. Life Sci. 2010, 67, 2215–2230. [Google Scholar] [CrossRef]
- Forbes, D.J.; Travesa, A.; Nord, M.S.; Bernis, C. Nuclear transport factors: Global regulation of mitosis. Curr. Opin. Cell Biol. 2015, 35, 78–90. [Google Scholar] [CrossRef]
- Kirli, K.; Karaca, S.; Dehne, H.J.; Samwer, M.; Pan, K.T.; Lenz, C.; Urlaub, H.; Görlich, D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. eLife 2015, 4, e11466. [Google Scholar] [CrossRef]
- Torosantucci, L.; De Luca, M.; Guarguaglini, G.; Lavia, P.; Degrassi, F. Localized RanGTP accumulation promotes microtubule nucleation at kinetochores in somatic mammalian cells. Mol. Biol. Cell. 2008, 19, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jiang, Q.; Zhang, C. A fraction of Crm1 locates at centrosomes by its CRIME domain and regulates the centrosomal localization of pericentrin. Biochem. Biophys. Res. Commun. 2009, 384, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.C.; Weiss, J.; Pham, N.A.; Li, Q.; Martins-Filho, S.N.; Wang, Y.; Tsao, M.S.; Moghal, N. Antitumor efficacy of XPO1 inhibitor Selinexor in KRAS-mutant lung adenocarcinoma patient-derived xenografts. Transl. Oncol. 2021, 14, 101179. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, D.J.; Finetti, P.; Birnbaum, D.; Mamessier, E.; Bertucci, F. XPO1 expression is a poor-prognosis marker in pancreatic adenocarcinoma. J. Clin. Med. 2019, 8, 596. [Google Scholar] [CrossRef]
- Zhao, L.; Luo, B.; Wang, L.; Chen, W.; Jiang, M.; Zhang, N. Pan-cancer analysis reveals the roles of XPO1 in predicting prognosis and tumorigenesis. Transl. Cancer Res. 2021, 10, 4664–4679. [Google Scholar] [CrossRef]
- Martignetti, J.; Razak, A.R.A.; Chen, Y.; Gabrail, N.Y.; Gericitano, J.F.; Camacho, C.; Pereira, E.; Evans, B.; Dottino, P.; McCauley, D.; et al. Preclinical and Early Clinical Activity of the Oral Selective Inhibitor of Nuclear Export (Sine) Exportin 1 (Xpo1) Antagonist Selinexor (Kpt-330) in Patients (Pts) with Platinum Resistant/Refractory Ovarian Cancer (Ovca). Ann. Oncol. 2014, 32 (Suppl. S15), 310. [Google Scholar] [CrossRef]
- Wang, A.Y.; Liu, H. The past, present, and future of CRM1/XPO1 inhibitors. Stem Cell Investig. 2019, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85. [Google Scholar] [CrossRef]
- Green, A.L.; Ramkissoon, S.H.; McCauley, D.; Jones, K.; Perry, J.A.; Hsu, J.H.; Ramkissoon, L.A.; Maire, C.L.; Hubbell-Engler, B.; Knoff, D.S.; et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015, 17, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Madduri, D.; Richard, S.; Chari, A. Selinexor in relapsed/refractory multiple myeloma. Ther. Adv. Hematol. 2020, 11, 2040620720930629. [Google Scholar] [CrossRef]
- Hays, J.; Zhang, J.; Berlin, J.D.; O’Hara, M.; Shah, M.A.; Reichmann, W.; Senapedis, W.; Achour, H.; Baloglu, E.; Shacham, S.; et al. Eltanexor (KPT-8602), a second-generation selective inhibitor of nuclear export (SINE) compound, in patients with metastatic colorectal cancer (mCRC). Ann. Oncol. 2018, 29, iii716. [Google Scholar] [CrossRef]
- Cornell, R.F.; Baz, R.; Richter, J.R.; Rossi, A.; Vogl, D.T.; Chen, C.; Shustik, C.; Alvarez, M.J.; Shen, Y.; Unger, T.J.; et al. A phase 1 clinical trial of oral Eltanexor in patients with relapsed or refractory multiple myeloma. Am. J. Hematol. 2022, 97, e54–e58. [Google Scholar] [CrossRef]
- Lee, S.; Bhatnagar, B.; Mohan, S.R.; William, S.T., Jr.; Baloglu, E.; Wang, H.; Shah, J.J.; Shacham, S.; Kauffman, M.G. Eltanexor (KPT-8602), a Second-Generation Selective Inhibitor of Nuclear Export (SINE) Compound, in Patients with Higher-Risk Myelodysplastic Syndrome. Blood 2019, 34 (Suppl. S1), 2997. [Google Scholar] [CrossRef]
- Lock, R.B.; Evans, K.; Jones, C.D.; Erickson, S.W.; Teicher, B.A.; Unger, T.J.; Landesman, Y.; Smith, M.A. Abstract 4181: The XPO1 inhibitor, Eltanexor, exhibits potent in vivo activity against a broad range of pediatric acute lymphoblastic leukemia subtypes. Cancer Res. 2020, 80 (Suppl. S16), 4181. [Google Scholar] [CrossRef]
- Lassman, A.B.; Wen, P.Y.; Van Den Bent, M.J.; Plotkin, S.R.; Walenkamp, A.M.E.; Huang, X.; Rodriguez-Lopez, K.; Kauffman, M.G.; Shacham, S.; Mau-Sørensen, M. Efficacy and safety of selinexor in recurrent glioblastoma. J. Clin. Oncol. 2019, 37 (Suppl. S15), 2005. [Google Scholar] [CrossRef]
- Lassman, A.B.; Wen, P.Y.; Van Den Bent, M.J.; Plotkin, S.R.; Walenkamp, A.M.E.; Green, A.L.; Li, K.; Walker, C.J.; Chang, H.; Tamir, S.; et al. A Phase II Study of the Efficacy and Safety of Oral Selinexor in Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 452–460. [Google Scholar] [CrossRef]
- Hannen, R.; Selmansberger, M.; Hauswald, M.; Pagenstecher, A.; Nist, A.; Stiewe, T.; Acker, T.; Carl, B.; Nimsky, C.; Bartsch, J.W. Comparative Transcriptomic Analysis of Temozolomide Resistant Primary GBM Stem-Like Cells and Recurrent GBM Identifies Up-Regulation of the Carbonic Anhydrase CA2 Gene as Resistance Factor. Cancers 2019, 11, 921. [Google Scholar] [CrossRef]
- Thome, I.; Lacle, R.; Voß, A.; Bortolussi, G.; Pantazis, G.; Schmidt, A.; Conrad, C.; Jacob, R.; Timmesfeld, N.; Bartsch, J.W.; et al. Neoplastic Cells are the Major Source of MT-MMPs in IDH1-Mutant Glioma, Thus Enhancing Tumor-Cell Intrinsic Brain Infiltration. Cancers 2020, 12, 2456. [Google Scholar] [CrossRef]
- Deshpande, K.; Saatian, B.; Martirosian, V.; Lin, M.; Julian, A.; Neman, J. Isolation of Neural Stem Cells from Whole Brain Tissues of Adult Mice. Curr. Protoc. Stem Cell Biol. 2019, 49, e80. [Google Scholar] [CrossRef]
- Cook, L.; Sengelmann, M.; Winkler, B.; Nagl, C.; Koch, S.; Schlomann, U.; Slater, E.P.; Miller, M.A.; von Strandmann, E.P.; Dörsam, B.; et al. ADAM8-Dependent Extracellular Signaling in the Tumor Microenvironment Involves Regulated Release of Lipocalin 2 and MMP-9. Int. J. Mol. Sci. 2022, 23, 1976. [Google Scholar] [CrossRef]
- Xia, M.; Knezevic, D.; Vassilev, L.T. p21 does not protect cancer cells from apoptosis induced by nongenotoxic p53 activation. Oncogene 2011, 30, 346–355. [Google Scholar] [CrossRef]
- Solozobova, V.; Rolletschek, A.; Blattner, C. Nuclear accumulation and activation of p53 in embryonic stem cells after DNA damage. BMC Cell Biol. 2009, 10, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, S.R.; Lopes, J.; Levin, N.K.; Kalpage, H.; Tainsky, M.A. Germline mutations in apoptosis pathway genes in ovarian cancer; the functional role of a TP53I3 (PIG3) variant in ROS production and DNA repair. Cell Death Discov. 2021, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Wahba, A.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The XPO1 Inhibitor Selinexor Inhibits Translation and Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and In Vivo. Mol. Cancer Ther. 2018, 17, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S.; et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef]
- Cornell, R.F.; Rossi, A.C.; Baz, R.; Hofmeister, C.C.; Shustik, C.; Richter, J.R.; Chen, C.I.; Vogl, D.T.; Baloglu, E.; Senapedis, W.; et al. Eltanexor (KPT-8602), a Second-Generation Selective Inhibitor of Nuclear Export (SINE) Compound, in Patients with Refractory Multiple Myeloma. Blood 2017, 130 (Suppl. S1), 3134. [Google Scholar] [CrossRef]
- Hing, Z.A.; Fung, H.Y.; Ranganathan, P.; Mitchell, S.; El-Gamal, D.; Woyach, J.A.; Williams, K.; Goettl, V.M.; Smith, J.; Yu, X.; et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016, 30, 2364–2372. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Cha, G.D.; Kang, T.; Baik, S.; Kim, D.; Choi, S.H.; Hyeon, T.; Kim, D.H. Advances in drug delivery technology for the treatment of glioblastoma multiforme. J. Control. Release 2020, 328, 350–367. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otte, K.; Zhao, K.; Braun, M.; Neubauer, A.; Raifer, H.; Helmprobst, F.; Barrera, F.O.; Nimsky, C.; Bartsch, J.W.; Rusch, T. Eltanexor Effectively Reduces Viability of Glioblastoma and Glioblastoma Stem-Like Cells at Nano-Molar Concentrations and Sensitizes to Radiotherapy and Temozolomide. Biomedicines 2022, 10, 2145. https://doi.org/10.3390/biomedicines10092145
Otte K, Zhao K, Braun M, Neubauer A, Raifer H, Helmprobst F, Barrera FO, Nimsky C, Bartsch JW, Rusch T. Eltanexor Effectively Reduces Viability of Glioblastoma and Glioblastoma Stem-Like Cells at Nano-Molar Concentrations and Sensitizes to Radiotherapy and Temozolomide. Biomedicines. 2022; 10(9):2145. https://doi.org/10.3390/biomedicines10092145
Chicago/Turabian StyleOtte, Katharina, Kai Zhao, Madita Braun, Andreas Neubauer, Hartmann Raifer, Frederik Helmprobst, Felipe Ovalle Barrera, Christopher Nimsky, Jörg W. Bartsch, and Tillmann Rusch. 2022. "Eltanexor Effectively Reduces Viability of Glioblastoma and Glioblastoma Stem-Like Cells at Nano-Molar Concentrations and Sensitizes to Radiotherapy and Temozolomide" Biomedicines 10, no. 9: 2145. https://doi.org/10.3390/biomedicines10092145
APA StyleOtte, K., Zhao, K., Braun, M., Neubauer, A., Raifer, H., Helmprobst, F., Barrera, F. O., Nimsky, C., Bartsch, J. W., & Rusch, T. (2022). Eltanexor Effectively Reduces Viability of Glioblastoma and Glioblastoma Stem-Like Cells at Nano-Molar Concentrations and Sensitizes to Radiotherapy and Temozolomide. Biomedicines, 10(9), 2145. https://doi.org/10.3390/biomedicines10092145