
Hepatitis B x (HBx) as a Component of a Functional Cure for Chronic Hepatitis B

Abstract
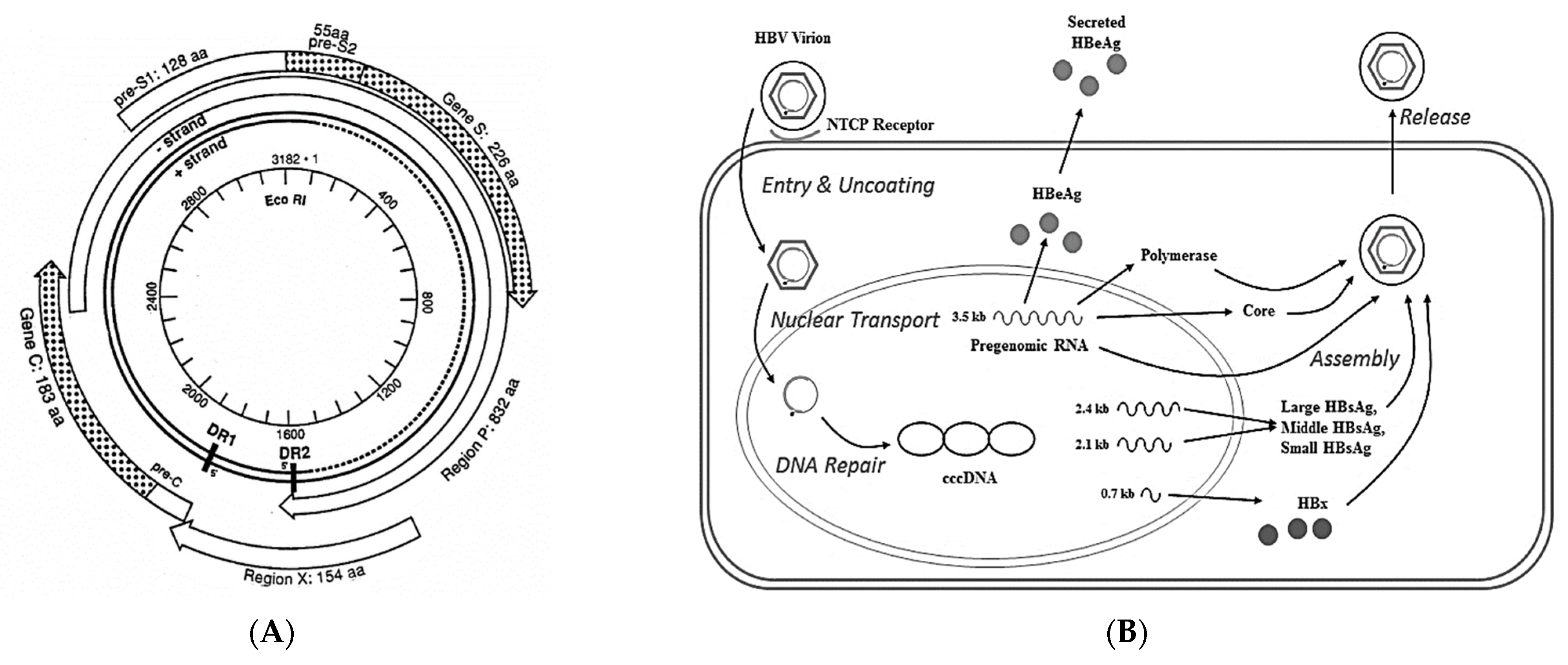
:1. Introduction

2. HBV Treatments and Their Limitations in Achieving a Functional Cure (Table 1)

{kind=link}
{kind=link}
{kind=link}
| Drug * | Mechanism of Action |
|---|---|
| Intron A, Pegasys | Immunostimulation with interferons |
| Epivir, Hepsera, Baraclude, Tyzeka, Viread, Vemlidy, Levovir, Besivo | Nucleoside analogs that inhibit the HBV polymerase |
| VIR-2218, RG6346, JNJ-3989, AB-729 | Inhibitory RNAs blocking HBV gene expression |
| Vebicorvir, Morphothiadin, JNJ 56136379, EDP-514, R07049389, QL-007, ABI-H3733, ZM-H1506R, ALG-000184, B-836, VNRX-9945 | Inhibitors that disrupt nucleocapsids or prevent normal nucleocapsid formation |
| NASVAC, GS-4774, HepTcell, VBI-2601, VVX001, VTP-300, CVI-HBV-002, AIC 649, HB-110, JNJ 64300535, CARG-201 | Therapeutic vaccines to target immunological resolution of the carrier state |
| Selgantolimod, RG7854 | Toll-like receptor agonists to stimulate innate immunity |
| Lenvervimab, Vir-3434 | Monoclonal antibodies against HBV antigens |
| ASX22, GS 4224 | PD-L1 inhibitors to overcome T cell exhaustion |
| Bulevirtide, Hepcludex, hzVSF | Inhibitors of virus entry into susceptible cells |
3. Role of HBx in the Immunopathogenesis of CLD and HCC
3.1. HBx and Mitochondria
3.2. HBx and Fibrogenesis
4. HBx and cccHBV DNA as Targets for a Functional Cure
5. HBx Integration
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α2M | alpha 2 macroglobulin |
| αSMA | alpha-smooth muscle actin |
| Ca2+ | calcium ions |
| ccc HBV DNA | covalently closed circular hepatitis B virus DNA |
| CLD | chronic liver disease |
| CHB | chronic hepatitis B |
| CTGF | connective-tissue growth factor |
| DAMPs | damage-associated molecular patterns |
| DR | direct repeats |
| ECM | extracellular matrix |
| ER | endoplasmic reticulum |
| FAK | focal activated kinase |
| HBeAg | hepatitis B e antigen |
| HBsAg | hepatitis B surface antigen |
| HBV | hepatitis B virus |
| HBx | hepatitis B x antigen |
| HCC | hepatocellular carcinoma |
| HIF-1α | hypoxia inducible factor 1α |
| IFN | interferon |
| LASP-1 | LIM and SH3 domain protein 1 |
| MAPK | mitogen-activated protein kinase |
| MAVS | mitochondrial antiviral signaling protein, matrix metalloproteinase-2 |
| NF-κB | nuclear factor kappa B |
| NTCP | Sodium taurocholate co-transporting polypeptide |
| ORF | open reading frame |
| pgRNA | pre-genomic RNA |
| PI3K | phosphoinositol 3-kinase |
| PRMT1 | protein arginine methyltransferase |
| Pyk2 | proline-rich tyrosine kinase 2 |
| RIG-1 | retinoic acid-inducible gene 1 |
| ROI | reactive oxygen intermediates |
| SCFAs | short-chain fatty acids |
| Smc | structural maintenance of chromosomes |
| TGFβ | transforming growth factor β |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor alpha |
| siRNAs | small interfering RNAs |
| WDR77 | WD repeat domain 77 protein |
| WHV | woodchuck hepatitis virus |
References
- Harputluoglu, M.; Carr, B.I. Hepatitis B Before and After Hepatocellular Carcinoma. J. Gastrointest. Cancer 2021, 52, 1206–1210. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Barreiro, P.; Cachay, E.; Kottilil, S.; Fernandez-Montero, J.V.; de Mendoza, C. Advances in hepatitis B therapeutics. Ther. Adv. Infect. Dis. 2020, 7, 2049936120965027. [Google Scholar] [CrossRef] [PubMed]
- Chien, R.N.; Liaw, Y.F. Current Trend in Antiviral Therapy for Chronic Hepatitis B. Viruses 2022, 14, 434. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2022, 22, 19–32. [Google Scholar] [CrossRef]
- Cacoub, P.; Terrier, B. Hepatitis B-related autoimmune manifestations. Rheum. Dis. Clin. N. Am. 2009, 35, 125–137. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Bonamassa, B.; Arzumanyan, A. The roles of hepatitis B virus-encoded X protein in virus replication and the pathogenesis of chronic liver disease. Expert Opin. Ther. Targets 2014, 18, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Garces, R.; Richardson, C.D. X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev. 2001, 12, 189–205. [Google Scholar] [CrossRef]
- Liu, S.; Koh, S.S.; Lee, C.G. Hepatitis B Virus X Protein and Hepatocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 940. [Google Scholar] [CrossRef] [PubMed]
- Wollersheim, M.; Debelka, U.; Hofschneider, P.H. A transactivating function encoded in the hepatitis B virus X gene is conserved in the integrated state. Oncogene 1988, 3, 545–552. [Google Scholar]
- Moini, M.; Fung, S. HBsAg Loss as a Treatment Endpoint for Chronic HBV Infection: HBV Cure. Viruses 2022, 14, 657. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.A.; Calabrese, D.; Suslov, A.; Terracciano, L.M.; Heim, M.H.; Wieland, S. Ubiquitous expression of HBsAg from integrated HBV DNA in patients with low viral load. J. Hepatol. 2021, 75, 840–847. [Google Scholar] [CrossRef] [PubMed]
- McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Merali, S.; Merali, C.; Feitelson, M. Short chain fatty acids delay the development of hepatocellular carcinoma in HBx transgenic mice. Neoplasia 2021, 23, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Ullrich, S.J.; Gangemi, J.D.; Kappel, C.A.; Ngo, L.; Feitelson, M.A.; Jay, G. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat. Genet. 1995, 9, 41–47. [Google Scholar] [CrossRef]
- Beasley, R.P.; Hwang, L.Y.; Lin, C.C.; Chien, C.S. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22707 men in Taiwan. Lancet 1981, 2, 1129–1133. [Google Scholar] [CrossRef]
- Mason, W.S.; Jilbert, A.R.; Litwin, S. Hepatitis B Virus DNA Integration and Clonal Expansion of Hepatocytes in the Chronically Infected Liver. Viruses 2021, 13, 210. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Larkin, J.D. New Animal Models of Hepatitis B and C. ILAR J. 2001, 42, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.K.K.; Andrisani, O. Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells. Genes 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Lok, A. Hepatitis B therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic Hepatitis B Infection: A Review. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.hepb.org/treatment-and-management/drug-watch/ (accessed on 16 August 2022).
- Dienstag, J.L.; Schiff, E.R.; Wright, T.L.; Perrillo, R.P.; Hann, H.W.; Goodman, Z.; Crowther, L.; Condreay, L.D.; Woessner, M.; Rubin, M.; et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N. Engl. J. Med. 1999, 341, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.; Wang, H.; Niu, J.; Chim, A.M.; Sung, J.J. Two-year lamivudine treatment for hepatitis B e antigen-negative chronic hepatitis B: A double-blind, placebo-controlled trial. Antivir. Ther. 2007, 12, 345–353. [Google Scholar] [CrossRef]
- Marcellin, P.; Chang, T.T.; Lim, S.G.; Sievert, W.; Tong, M.; Arterburn, S.; Borroto-Esoda, K.; Frederick, D.; Rousseau, F. Long-term efficacy and safety of adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. Hepatology 2008, 48, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Tassopoulos, N.C.; Heathcote, E.J.; Chang, T.T.; Kitis, G.; Rizzetto, M.; Marcellin, P.; Lim, S.G.; Goodman, Z.; Ma, J.; et al. Adefovir Dipivoxil 438 Study Group. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology 2006, 131, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cai, D.C.; Hu, P.; Ren, H. Low-level viremia in nucleoside analog-treated chronic hepatitis B patients. Chin. Med. J. 2021, 134, 2810–2817. [Google Scholar] [CrossRef] [PubMed]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. Toll-Like Receptor Response to Hepatitis B Virus Infection and Potential of TLR Agonists as Immunomodulators for Treating Chronic Hepatitis B: An Overview. Int. J. Mol. Sci. 2021, 22, 10462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 2010, 1, 1106–1117. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Wei, L.; PLoss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. [Google Scholar] [CrossRef]
- Tang, L.; Zhao, Q.; Wu, S.; Cheng, J.; Chang, J.; Guo, J.T. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin. Drug Discov. 2017, 12, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.W.; Mak, L.Y.; Seto, W.K.; Yuen, M.F. RNA interference as a novel treatment strategy for chronic hepatitis B infection. Clin. Mol. Hepatol. 2022, in press. [CrossRef] [PubMed]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ko, C.; Lee, J.Y.; Kim, M. Current Progress in the Development of Hepatitis B Virus Capsid Assembly Modulators: Chemical Structure, Mode-of-Action and Efficacy. Molecules 2021, 26, 7420. [Google Scholar] [CrossRef]
- Moyo, B.; Bloom, K.; Scott, T.; Ely, A.; Arbuthnot, P. Advances with using CRISPR/Cas-mediated gene editing to treat infections with hepatitis B virus and hepatitis C virus. Virus Res. 2018, 244, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Kostyushev, D.; Brezgin, S.; Kostyusheva, A.; Zarifyan, D.; Goptar, I.; Chulanov, V. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell Mol. Life Sci. 2019, 76, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Schiwon, M.; Ehrke-Schulz, E.; Oswald, A.; Bergmann, T.; Michler, T.; Protzer, U.; Ehrhardt, A. One-Vector System for Multiplexed CRISPR/Cas9 against Hepatitis B Virus cccDNA Utilizing High-Capacity Adenoviral Vectors. Mol. Ther. Nucleic Acids 2018, 12, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. CRISPR/Cas9-Based Antiviral Strategy: Current Status and the Potential Challenge. Molecules 2019, 24, 1349. [Google Scholar] [CrossRef] [PubMed]
- Prescott, N.A.; Bram, Y.; Schwartz, R.E.; David, Y. Targeting Hepatitis B Virus Covalently Closed Circular DNA and Hepatitis B Virus X Protein: Recent Advances and New Approaches. ACS Infect. Dis. 2019, 5, 1657–1667. [Google Scholar] [CrossRef]
- Hurwitz, S.J.; McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Tao, S.; Bassit, L.; Chen, Z.; Kohler, J.J.; Amblard, F.; Feitelson, M.A.; et al. Studies on the Efficacy, Potential Cardiotoxicity and Monkey Pharmacokinetics of GLP-26 as a Potent Hepatitis B Virus Capsid Assembly Modulator. Viruses 2021, 13, 114. [Google Scholar] [CrossRef]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Yuen Chan, H.L.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Cheng, Y.S.; Lo, D.C.; Zheng, W. Drug combination therapy for emerging viral diseases. Drug Discov. Today 2021, 26, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Medhat, A.; Arzumanyan, A.; Feitelson, M.A. Hepatitis B x antigen (HBx) is an important therapeutic target in the pathogenesis of hepatocellular carcinoma. Oncotarget 2021, 12, 2421–2433. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Weng, C.H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Clayton, M.; Sun, B.; Perchonock, C.E.; Morgan, J.L.; Siracusa, L.D.; Michaels, F.H.; Feitelson, M.A. Hepatitis B virus transgenic mouse model of chronic liver disease. Nat. Med. 1999, 5, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Fioravanti, J.; Di Lucia, P.; Magini, D.; Moalli, F.; Boni, C.; Benechet, A.P.; Fumagalli, V.; Inverso, D.; Vecchi, A.; Fiocchi, A.; et al. Effector CD8+ T cell-derived interleukin-10 enhances acute liver immunopathology. J. Hepatol. 2017, 67, 543–548. [Google Scholar] [CrossRef]
- Wieland, S.F.; Spangenberg, H.C.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc. Natl. Acad. Sci. USA 2004, 101, 2129–2134. [Google Scholar] [CrossRef]
- Mason, W.S.; Litwin, S.; Xu, C.; Jilbert, A.R. Hepatocyte turnover in transient and chronic hepadnavirus infections. J. Viral Hepat. 2007, 14, 22–28. [Google Scholar] [CrossRef]
- Murray, J.M.; Wieland, S.F.; Purcell, R.H.; Chisari, F.V. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. USA 2005, 102, 17780–17785. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N.; Nagaoka, T.; Yamashiro, M.; Mochizuki, K.; Kaneko, A.; Yamamoto, K.; Omura, M.; Hikiji, K.; Kato, M. Long-term histologic and virologic outcomes of acute self-limited hepatitis B. Hepatology 2003, 37, 1172–1179. [Google Scholar] [CrossRef]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, G.; Caccamo, G.; Filomia, R.; Pollicino, T. Occult HBV infection. Semin. Immunopathol. 2013, 35, 39–52. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999, 284, 825–829. [Google Scholar] [CrossRef]
- Datfar, T.; Doulberis, M.; Papaefthymiou, A.; Hines, I.N.; Manzini, G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens 2021, 10, 1366. [Google Scholar] [CrossRef]
- Yang, H.I.; Lu, S.N.; Liaw, Y.F.; You, S.L.; Sun, C.A.; Wang, L.Y.; Hsiao, C.K.; Chen, P.J.; Chen, D.S.; Chen, C.J.; et al. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N. Engl. J. Med. 2002, 347, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, L.Y.; Zhang, S.; Qiu, L.Y.; Li, N.; Zhang, X.; Zhang, X.Z.; Shan, C.L.; Ye, L.H.; Zhang, X.D. Anti-hepatitis B virus X protein in sera is one of the markers of development of liver cirrhosis and liver cancer mediated by HBV. J. Biomed. Biotechnol. 2009, 2009, 289068. [Google Scholar] [CrossRef]
- Chun, E.; Lee, J.; Cheong, H.S.; Lee, K.Y. Tumor eradication by hepatitis B virus X antigen-specific CD8+ T cells in xenografted nude mice. J. Immunol. 2003, 170, 1183–1190. [Google Scholar] [CrossRef]
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Sivasudhan, E.; Blake, N.; Lu, Z.; Meng, J.; Rong, R. Hepatitis B Viral Protein HBx and the Molecular Mechanisms Modulating the Hallmarks of Hepatocellular Carcinoma: A Comprehensive Review. Cells 2022, 11, 741. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. Corrigendum to “EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2019, 70, 817. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–6811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J.; Lab for Trauma and Surgical Infections. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Waris, G.; Huh, K.W.; Siddiqui, A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol. Cell. Biol. 2001, 21, 7721–7730. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Han, D.D.; Wright, C.J.; Burch, T.; Piper, J.; Osiowy, C.; Gao, C.; Chiang, S.; Magill, T.; Dick, K.; et al. The interaction between hepatitis B virus X protein and AIB1 oncogene is required for the activation of NFκB signal transduction. Biochem. Biophys. Res. Commun. 2012, 423, 6–12. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Reis, H.M.; Tufan, N.L.; Sun, B.; Pan, J.; Lian, Z. Putative roles of hepatitis B x antigen in the pathogenesis of chronic liver disease. Cancer Lett. 2009, 286, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Siddiqui, A. Hepatitis B virus X protein stimulates the mitochondrial translocation of Raf-1 via oxidative stress. J. Virol. 2007, 81, 6757–6760. [Google Scholar] [CrossRef]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef]
- Murata, M.; Matsuzaki, K.; Yoshida, K.; Sekimoto, G.; Tahashi, Y.; Mori, S.; Uemura, Y.; Sakaida, N.; Fujisawa, J.; Seki, T.; et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology 2009, 49, 1203–1217. [Google Scholar] [CrossRef]
- Martín-Vílchez, S.; Sanz-Cameno, P.; Rodríguez-Muñoz, Y.; Majano, P.L.; Molina-Jiménez, F.; López-Cabrera, M.; Moreno-Otero, R.; Lara-Pezzi, E. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology 2008, 47, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.H.; Tan, D.M.; Zhu, P.A.; Liu, F. Hepatitis B virus X protein promotes proliferation and upregulates TGF-beta1 and CTGF in human hepatic stellate cell line, LX-2. Hepatobiliary Pancreat. Dis. Int. 2009, 8, 59–64, PMID: 19208517. [Google Scholar] [PubMed]
- Lee, D.K.; Park, S.H.; Yi, Y.; Choi, S.G.; Lee, C.; Parks, W.T.; Cho, H.; de Caestecker, M.P.; Shaul, Y.; Roberts, A.B.; et al. The hepatitis B virus encoded oncoprotein pX amplifies TGF-beta family signaling through direct interaction with Smad4: Potential mechanism of hepatitis B virus-induced liver fibrosis. Genes Dev. 2001, 15, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Chen, M.; Mo, H.H.; Tsai, W.C.; Chang, Y.C.; Chang, C.C.; Chen, K.C.; Wu, H.Y.; Yuan, C.H.; Lee, C.H.; et al. Utilizing Experimental Mouse Model to Identify Effectors of Hepatocellular Carcinoma Induced by HBx Antigen. Cancers 2020, 12, 409. [Google Scholar] [CrossRef]
- Norton, P.A.; Reis, H.M.; Prince, S.; Larkin, J.; Pan, J.; Liu, J.; Gong, Q.; Zhu, M.; Feitelson, M.A. Activation of fibronectin gene expression by hepatitis B virus x antigen. J. Viral Hepat. 2004, 11, 332–341. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Kammerer, U.; Winkler, C.; Schindler, D.; Sickmann, A.; Honig, A.; Butt, E. Overexpression of LASP-1 mediates migration and proliferation of human ovarian cancer cells and influences zyxin localisation. Br. J. Cancer 2007, 96, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Katrinli, S.; Ozdil, K.; Sahin, A.; Ozturk, O.; Kir, G.; Baykal, A.T.; Akgun, E.; Sarac, O.S.; Sokmen, M.; Doğanay, H.L.; et al. Proteomic profiling of HBV infected liver biopsies with different fibrotic stages. Proteome Sci. 2017, 15, 7. [Google Scholar] [CrossRef]
- Kongkavitoon, P.; Tangkijvanich, P.; Hirankarn, N.; Palaga, T. Hepatitis B Virus HBx Activates Notch Signaling via Delta-Like 4/Notch1 in Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0146696. [Google Scholar] [CrossRef]
- Yang, S.L.; Ren, Q.G.; Zhang, T.; Pan, X.; Wen, L.; Hu, J.L.; Yu, C.; He, Q.J. Hepatitis B virus X protein and hypoxia-inducible factor-1α stimulate Notch gene expression in liver cancer cells. Oncol. Rep. 2017, 37, 348–356. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol. Res. 2016, 108, 57–64. [Google Scholar] [CrossRef]
- Tschaharganeh, D.F.; Chen, X.; Latzko, P.; Malz, M.; Gaida, M.M.; Felix, K.; Ladu, S.; Singer, S.; Pinna, F.; Gretz, N.; et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 2013, 144, 1530–1542.e12. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, C.; Hong, L.; Wang, J.; Du, Y.; Song, J.; Shao, X.; Zhang, J.; Han, H.; Liu, J.; et al. Expression of Jagged1 and its association with hepatitis B virus X protein in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2007, 356, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Clayton, M.M. X antigen polypeptides in the sera of hepatitis B virus-infected patients. Virology 1990, 177, 367–371. [Google Scholar] [CrossRef]
- Horiike, N.; Blumberg, B.S.; Feitelson, M.A. Characteristics of hepatitis B X antigen, antibodies to X antigen, and antibodies to the viral polymerase during hepatitis B virus infection. J. Infect. Dis. 1991, 164, 1104–1112. [Google Scholar] [CrossRef]
- Hwang, G.Y.; Lin, C.Y.; Huang, L.M.; Wang, Y.H.; Wang, J.C.; Hsu, C.T.; Yang, S.S.; Wu, C.C. Detection of the hepatitis B virus X protein (HBx) antigen and anti-HBx antibodies in cases of human hepatocellular carcinoma. J. Clin. Microbiol. 2003, 41, 5598–5603. [Google Scholar] [CrossRef]
- Xu, Z.; Yen, T.S.; Wu, L.; Madden, C.R.; Tan, W.; Slagle, B.L.; Ou, J.H. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J. Virol. 2002, 76, 2579–2584. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Kaneko, S.; Girones, R.; Anderson, R.W.; Hornbuckle, W.E.; Tennant, B.C.; Cote, P.J.; Gerin, J.L.; Purcell, R.H.; Miller, R.H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol. 1993, 67, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Schirmacher, P.; Rogler, C.E. Woodchuck hepatitis virus X protein is present in chronically infected woodchuck liver and woodchuck hepatocellular carcinomas which are permissive for viral replication. J. Virol. 1996, 70, 5246–5254. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Duan, L.X.; Guo, J.; Sun, B.; Woo, J.; Steensma, K.; Horiike, N.; Blumberg, B.S. X region deletion variants of hepatitis B virus in surface antigen-negative infections and non-A, non-B hepatitis. J. Infect. Dis. 1995, 172, 713–722. [Google Scholar] [CrossRef]
- Carmona, S.; Ely, A.; Crowther, C.; Moolla, N.; Salazar, F.H.; Marion, P.L.; Ferry, N.; Weinberg, M.S.; Arbuthnot, P. Effective inhibition of HBV replication in vivo by anti-HBx short hairpin RNAs. Mol. Ther. 2006, 13, 411–421. [Google Scholar] [CrossRef]
- Malmassari, S.; Lone, Y.C.; Zhang, M.; Transy, C.; Michel, M.L. In vivo hierarchy of immunodominant and subdominant HLA-A*0201-restricted T-cell epitopes of HBx antigen of hepatitis B virus. Microbes Infect. 2005, 7, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Yoon, H.; Min, S.S.; Lee, H.G.; Kim, Y.J.; Lee, T.G.; Lim, J.S.; Kim, C.M.; Park, S.N. Induction of cytotoxic T lymphocytes with peptides in vitro: Identification of candidate T-cell epitopes in hepatitis B virus X antigen. J. Immunother. 1999, 22, 279–287. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Yang, G. Research progress in hepatitis B virus covalently closed circular DNA. Cancer Biol. Med. 2021, 19, 415–431. [Google Scholar] [CrossRef]
- Ma, J.; Sun, T.; Park, S.; Shen, G.; Liu, J. The role of hepatitis B virus X protein is related to its differential intracellular localization. Acta Biochim. Biophys. Sin. 2011, 43, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.; Zhang, F.; Liu, X.; Qin, S.; Yang, X.; Kong, D.; Pan, X.; You, H.; Zheng, K.; Tang, R. Calcium signaling in hepatitis B virus infection and its potential as a therapeutic target. Cell Commun. Signal. 2021, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Park, Y.K.; Shin, C.Y.; Park, S.H.; Ahn, S.H.; Kim, D.H.; Lim, K.H.; Kwon, S.Y.; Kim, K.P.; Yang, S.I.; et al. Hepatitis B virus inhibits liver regeneration via epigenetic regulation of urokinase-type plasminogen activator. Hepatology 2013, 58, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Tralhao, J.G.; Roudier, J.; Morosan, S.; Giannini, C.; Tu, H.; Goulenok, C.; Carnot, F.; Zavala, F.; Joulin, V.; Kremsdorf, D.; et al. Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: An alternative mechanism of HBx-related pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 6991–6996. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef]
- Benhenda, S.; Ducroux, A.; Rivière, L.; Sobhian, B.; Ward, M.D.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.L.; Benkirane, M.; et al. Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef]
- Yuan, H.; Zhao, L.; Yuan, Y.; Yun, H.; Zheng, W.; Geng, Y.; Yang, G.; Wang, Y.; Zhao, M.; Zhang, X. HBx represses WDR77 to enhance HBV replication by DDB1-mediated WDR77 degradation in the liver. Theranostics 2021, 11, 8362–8378. [Google Scholar] [CrossRef] [PubMed]
- Decorsière, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef]
- Pollicino, T.; Caminiti, G. HBV-Integration Studies in the Clinic: Role in the Natural History of Infection. Viruses 2021, 13, 368. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef]
- Wang, W.L.; London, W.T.; Lega, L.; Feitelson, M.A. HBxAg in the liver from carrier patients with chronic hepatitis and cirrhosis. Hepatology 1991, 14, 29–37. [Google Scholar] [CrossRef]
- Wang, W.L.; London, W.T.; Feitelson, M.A. Hepatitis B x antigen in hepatitis B virus carrier patients with liver cancer. Cancer Res. 1991, 51, 4971–4977. [Google Scholar] [PubMed]
- Martin-Vilchez, S.; Lara-Pezzi, E.; Trapero-Marugán, M.; Moreno-Otero, R.; Sanz-Cameno, P. The molecular and pathophysiological implications of hepatitis B X antigen in chronic hepatitis B virus infection. Rev. Med. Virol. 2011, 21, 315–329. [Google Scholar] [CrossRef]
- Guo, S.P.; Wang, W.L.; Zhai, Y.Q.; Zhao, Y.L. Expression of nuclear factor-kappa B in hepatocellular carcinoma and its relation with the X protein of hepatitis B virus. World J. Gastroenterol. 2001, 7, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Caselmann, W.H.; Schlüter, V.; Schreck, R.; Hofschneider, P.H.; Baeuerle, P.A. Hepatitis B virus transactivator MHBst: Activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO J. 1992, 11, 2991–3001. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef]
- Yang, L.; Zou, T.; Chen, Y.; Zhao, Y.; Wu, X.; Li, M.; Du, F.; Chen, Y.; Xiao, Z.; Shen, J. Hepatitis B virus X protein mediated epigenetic alterations in the pathogenesis of hepatocellular carcinoma. Hepatol. Int. 2022, in press. [CrossRef] [PubMed]
- Pant, K.; Mishra, A.K.; Pradhan, S.M.; Nayak, B.; Das, P.; Shalimar, D.; Saraya, A.; Venugopal, S.K. Butyrate inhibits HBV replication and HBV-induced hepatoma cell proliferation via modulating SIRT-1/Ac-p53 regulatory axis. Mol. Carcinog. 2019, 58, 524–532. [Google Scholar] [CrossRef]
- Simon, T.G.; Duberg, A.S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N. Engl. J. Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kobayashi, M.; Someya, T.; Saitoh, S.; Hosaka, T.; Akuta, N.; Suzuki, F.; Suzuki, Y.; Arase, Y.; Kumada, H. Occult hepatitis B virus infection increases hepatocellular carcinogenesis by eight times in patients with non-B, non-C liver cirrhosis: A cohort study. J. Viral Hepat. 2009, 16, 437–443. [Google Scholar] [CrossRef]
- Paterlini, P.; Driss, F.; Nalpas, B.; Pisi, E.; Franco, D.; Berthelot, P.; Bréchot, C. Persistence of hepatitis B and hepatitis C viral genomes in primary liver cancers from HBsAg-negative patients: A study of a low-endemic area. Hepatology 1993, 17, 20–29, PMID: 8380790. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Gan, W.; Zhao, Q.; Yin, Y.; Gao, Z. Functional cure of chronic hepatitis B: Efforts and prospects. Liver Res. 2020, 4, 329–336. [Google Scholar] [CrossRef]
- Thimme, R.; Dandri, M. Dissecting the divergent effects of interferon-alpha on immune cells: Time to rethink combination therapy in chronic hepatitis B? J. Hepatol. 2013, 58, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kairuz, D.; Arbuthnot, P.; Bloom, K. Silencing hepatitis B virus covalently closed circular DNA: The potential of an epigenetic therapy approach. World J. Gastroenterol. 2021, 27, 3182–3207. [Google Scholar] [CrossRef]
- Minor, M.M.; Slagle, B.L. Hepatitis B virus HBx protein interactions with the ubiquitin proteasome system. Viruses 2014, 6, 4683–4702. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, W.; Song, J.; Wu, Y.; Ni, B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol. Cell. Biol. 2013, 33, 2810–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feitelson, M.A.; Arzumanyan, A.; Spector, I.; Medhat, A. Hepatitis B x (HBx) as a Component of a Functional Cure for Chronic Hepatitis B. Biomedicines 2022, 10, 2210. https://doi.org/10.3390/biomedicines10092210
Feitelson MA, Arzumanyan A, Spector I, Medhat A. Hepatitis B x (HBx) as a Component of a Functional Cure for Chronic Hepatitis B. Biomedicines. 2022; 10(9):2210. https://doi.org/10.3390/biomedicines10092210
Chicago/Turabian StyleFeitelson, Mark A., Alla Arzumanyan, Ira Spector, and Arvin Medhat. 2022. "Hepatitis B x (HBx) as a Component of a Functional Cure for Chronic Hepatitis B" Biomedicines 10, no. 9: 2210. https://doi.org/10.3390/biomedicines10092210