
The NF-κB Pharmacopeia: Novel Strategies to Subdue an Intractable Target

 , , ,
, , ,
Abstract
:1. Introduction
2. Therapeutic Targeting of the NF-κB Pathway in Cancer
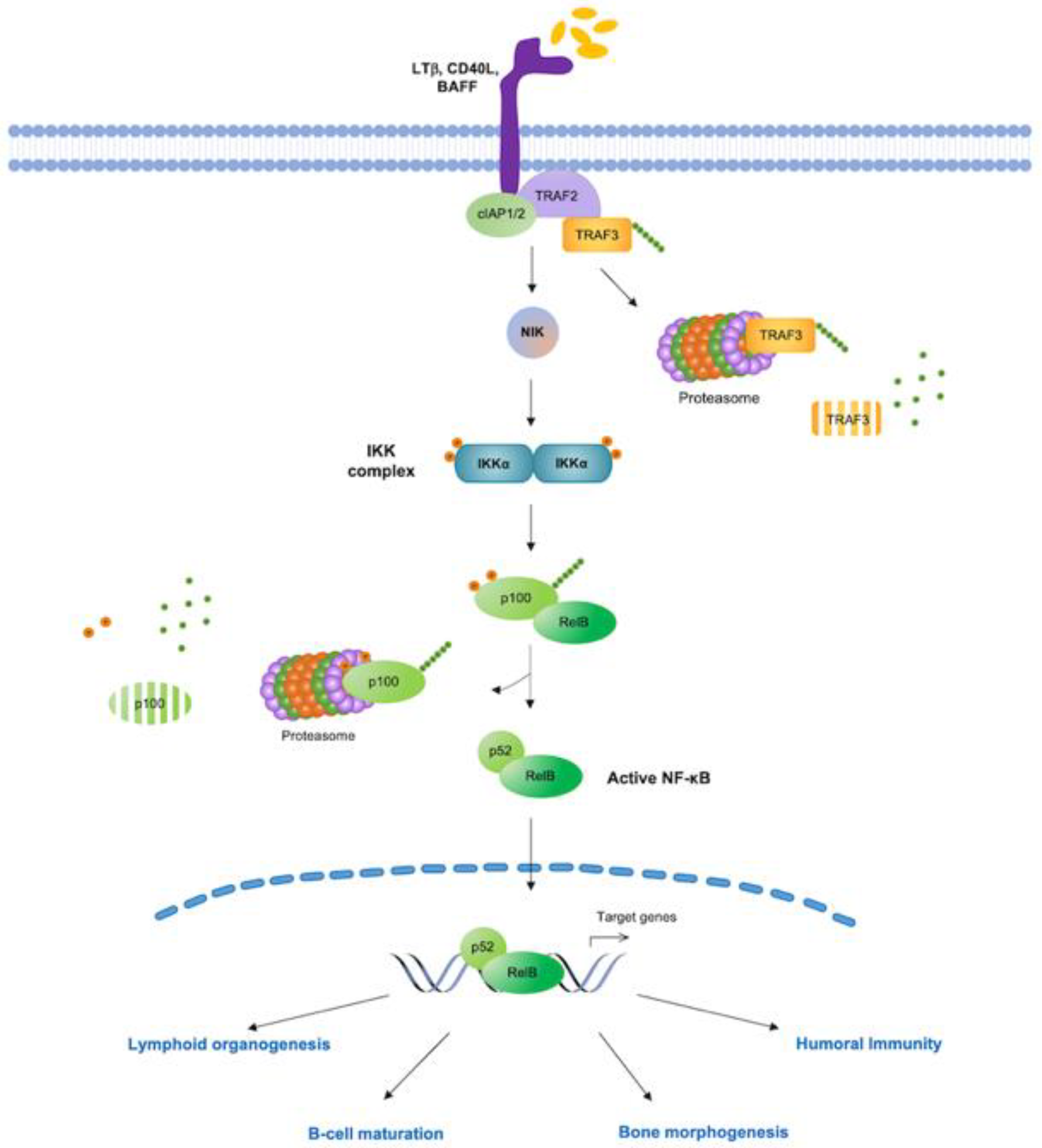
2.1. Agents Acting Upstream of IKK
2.1.1. TNF Receptors (TNF-Rs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Target | Cancer Type | Ongoing Clinical Trials | Phase | Recruitment Status | Other Information | Refs |
|---|---|---|---|---|---|---|---|
| Upstream IKKs complex | |||||||
| Brentuximab (Vedotin) | CD30 | HL; ALCL | NCT01657331 | 1–2 | Completed | Combination with bendamustine is safe and effective. Could be used as second-line therapy | [32] |
| ALK + ALCL | NCT02462538 | 1–2 | Terminated | ||||
| ASM; Mast Cell Leukemia; SM | NCT01807598 | 2 | Completed | BV is not active as a single agent in CD30+ advSM | [33] | ||
| HL; systemic ALCL | NCT02939014 | 2 | Completed | Positive benefit–risk profile for patients with R/R cHL and sALCL, confirming it as a potential treatment option | [34] | ||
| MMe | NCT03007030 | 2 | Recruiting | ||||
| PTCL; Paediatric HL | NCT02169505 | 2 | Terminated | Treatment performed after Allogeneic and Haploidentical Stem Cell Transplantation in High Risk CD30+ Lymphoma | |||
| Relapsed HL | NCT01900496 | 1 | Terminated | Combination therapy with Rituximab | |||
| Idelalisib (Cal-101) | PI3K | CLL | NCT01539291 | 3 | Terminated | Double-Blind extension study evaluating the efficacy and safety of different dose levels of single-agent Idelalisib | |
| R/R HL | NCT01393106 | 2 | Completed | [35] | |||
| FL; SLL | NCT02258529 | 2 | Terminated | Combination with rituximab in previously untreated adults with FL, SLL | |||
| Copansilib (BAY 80-6946) | PI3K | DBLCL | NCT04433182 | 2 | Recruiting | Combination regimen with rituximab-bendamustine | [36] |
| DLCBL | NCT04263584 | 2 | Recruiting | Combination with rituximab-CHOP in patients with untreated DLBCL | |||
| R/R MCL | NCT04939272 | 1–2 | Recruiting | Combination with venetoclax | |||
| MZL | NCT03474744 | 2 | Recruiting | Combination with rituximab | [37] | ||
| Ibrutinib (PCI-32765) | BTK | CLL | NCT02801578 | 2–3 | Completed | After one cycle at the prescribed 420 mg/d dose, ibrutinib dose can be reduced in subsequent cycles without loss of biological activity | [38] |
| High risk Smoldering MM | NCT02943473 | 2 | Terminated | ||||
| NSCLC | NCT02321540 | 1b-2 | Completed | ||||
| MCL | NCT02558816 | 1–2 | Active | The combination of obinutuzumab, ibrutinib, and venetoclax is well tolerated and provides high response rates, including at the molecular level, in relapsed and untreated MCL patients | [39] | ||
| Relapsed, Refractory, or High-Risk Untreated CLL; SLL; RS | NCT02420912 | 2 | Completed | The combination of nivolumab and ibrutinib has clinical activity in pts with RT with a 43% response rate | [40] | ||
| CLL | NCT02315768 | 1–2 | Active | Combination with obinutuzumab | |||
| Advanced FL | NCT02451111 | 2 | Active | Combination with rituximab | |||
| MCL | NCT02356458 | 1–2 | Terminated | Combination therapy with bortezomib followed by ibrutinib maintenance therapy | |||
| IMO-8400 | TLR 7, 8, and 9 | DLBCL | NCT02252146 | 1–2 | Completed | Dose-escalation study | |
| WM | NCT02092909 | 1–2 | Terminated | Lack of efficacy | |||
| WM | NCT02363439 | 1–2 | Completed | Lack of efficacy | |||
| LCL-161 | cIAPs | SCL; GM | NCT02649673 | 1 | Terminated | Combination with topotecan | |
| CRC; NSCLC; TNBC; RCC | NCT02890069 | 1 | Completed | Combination with several agents with immunomodulatory activity | |||
| R/R MM | NCT01955434 | 2 | Completed | Tested as single agent and in combination with cyclophosphamide | |||
| MM | NCT03111992 | 1 | Completed | Combination with CJM112, and PDR001 | |||
| Birinapant (TL32711) | cIAPs | Solid Tumors | NCT02587962 | 1–2 | Terminated | Combination with pembrolizumab | |
| High grade serum carcinoma | NCT02756130 | 1–2 | Withdrawn | Combination with platinum-based chemotherapy | |||
| R/R Solid Tumors | NCT04553692 | 1 | Recruiting | Combination with IGM-8444, Venetoclax, Bevacizumab, FOLFIRI, | |||
| Refractory Solid Tumors or Lymphoma | NCT00993239 | 1 | Completed | ||||
| NF-κB Core pathway | |||||||
| Icaritin | IKKα | HCC | NCT03236636 | 3 | Recruiting | Tested as single agent | [41] |
| HCC | NCT03236649 | 3 | Recruiting | Tested in PD-L1+ advanced HCC | [41] | ||
| Bortezomib | Proteasome | High-risk MM | NCT02308280 | 2 | Active | Following nonmyeloablative allogeneic transplant | |
| R/R ALL | NCT02535806 | 2 | Terminated | ||||
| AML | NCT01736943 | 2 | Completed | Combination with doxil/lipodox | |||
| AML | NCT01534260 | 1–2 | Completed | ||||
| R/R Lymphoma | NCT02613598 | 1 | Completed | Combination with ruxolitinib | |||
| MCL | NCT02356458 | 1–2 | Terminated | Combination with ibrutinib | |||
| MM | NCT01241708 | 3 | Active | ||||
| MCL | NCT03016988 | 2 | Unknown | Combination with fludarabine and cytarabine | |||
| Neuroblastoma | NCT02139397 | 1–2 | Active | Combination with DFMO | |||
| MM | NCT02237261 | 2 | Completed | ||||
| Carfizomib | Proteasome | MM | NCT02302495 | 2 | Active | ||
| MM | NCT02572492 | 2 | Active | ||||
| NET | NCT02318784 | 2 | Completed | ||||
| R/R NHL | NCT02142530 | 1 | Completed | Combination with belinostat | |||
| R/R NHL; R/R HL | NCT02867618 | 1–2 | Terminated | Combination with TGR-1202 | |||
| TCL | NCT01738594 | 1 | Terminated | Tested as single agent or combination with romidepsin | |||
| R/R Solid Tumors or Leukemia | NCT02512926 | 1 | Recruiting | Combination with cyclophosphamide and etoposide | |||
| Ixazomib (MNL-9708) | Proteasome | GBM | NCT02630030 | 1 | Completed | Orally administered ixazomib reaches brain tumor tissue. Therapeutic potential needs to be determined | [42] |
| MM, Lymphoma | NCT02924272 | 2 | Active | ||||
| Solid Tumors | NCT02942095 | 1 | Active | Combination with erlotinib | |||
| MM | NCT02312258 | 3 | Active | ||||
| MM | NCT02477215 | 1–2 | Completed | Tested as single agent or combination with bendamustine | |||
| B cell Lymphoma | NCT02898259 | 1–2 | Active | Combination with lenalidomide plus rituximab | |||
| MLN4924 (Pevonedistat) | NAE | AML | NCT01814826 | 1 | Completed | Combination with azacitidine | |
| AML; MDS | NCT02782468 | 1 | Completed | Tested as single agent and in combination with azacitidine | |||
| AML; MDS | NCT02610777 | 2 | Completed | ||||
| AML | NCT03009240 | 1 | Active | Combination with decitabine | |||
| AML | NCT04090736 | 3 | Recruiting | Tested as single agent and in combination with azacitidine | |||
| Solid Tumors | NCT03057366 | 1 | Completed | [43] | |||
| Vorinostat | HDAC | Solid Tumors | NCT04308330 | 1 | Recruiting | Combination with chemotherapy in R/R solid tumors | |
| Azacitidine | DNMT | Breast cancer | NCT04891068 | 2 | Recruiting | To determine the effect of low dose azacitidine therapy on tumor infiltrating lymphocytes (TILs) in primary tumors | |
| R/R Peripheral TCL | NCT05182957 | 2 | Recruiting | Combination with lenalidomide and anti-PD-1 monoclonal antibody | |||
| Decitabine | DNMT | Solid Tumors | NCT03875287 | 1 | Recruiting | Combination with cedazuridine | |
| NHL | NCT04697940 | 1–2 | Recruiting | Decitabine-primed CAR-T-cells in B-cell malignancies | |||
| Eltanexor (KPT-8602) | NE | R/R MM; metastatic CRC; metatstatic CRPC; HR-MDS | NCT02649790 | 1–2 | Recruiting | [44] | |
| Selinexor (KPT-330) | NE | Metastatic CRC | NCT04854434 | 2 | Active | Tested as single agent and in combination with pembrolizumab | |
| MM | NCT03110562 | 3 | Active | A once-per-week regimen of selinexor, bortezomib, and dexamethasone is a novel, effective, and convenient | [45] | ||
| ZEN003694 | BET | Metastatic CRPC | NCT04986423 | 2 | Recruiting | Combination with enzalutamide | |
| BMS-986158 | BET | Pediatric Cancer | NCT03936465 | 1 | Recruiting | ||
| NF-κB target genes | |||||||
| DTP3 | GADD45β/MKK7 | MM | MR/V027581/1 | 1–2 | Active | ||
| Venetoclax (ABT-199) | BCL-2 | WM | NCT02677324 | 2 | Completed | Venetoclax is safe and highly active in patients WM | [46] |
| MCL | NCT02471391 | 2 | Active | Combination with ibrutinib | [47] | ||
| MCL | NCT02558816 | 1–2 | Active | Combination with ibrutinib and Obinutuzumabis well tolerated and highly active | [39] | ||
| AML | NCT02203773 | 1 | Terminated | Combination with decitabine or azacytidine | [48] | ||
| NHL, DLBCL | NCT02055820 | 1–2 | Completed | Combination of with R-/G-CHOP in NHL demonstrated manageable safety and promising efficacy. Established a dose regimen for venetoclax plus R-CHOP in DLBCL | [49,50] | ||
| R/R DLBCL | NCT03136497 | 1 | Active | Combination with ibrutinib and rituximab | |||
| CLL | NCT03128879 | 2 | Recruiting | Combination with ibrutinib | |||
| R/R CLL | NCT02427451 | 1–2 | Active | Combination with obinutuzumab and ibrutinib | [51] | ||
| Navitoclax (ABT-737) | BCL-2 | Advanced or metastatic solid tumors | NCT02079740 | 1–2 | Recruiting | Combination with trametinib | |
| Advanced Myeloid Neoplasms | NCT05455294 | 1 | Recruiting | Combination with decitabine, and venetoclax | |||
| Bevacizumab | VEGF-A | HCC | NCT03434379 | 3 | Active | Combination with atezolizumab | [52] |
| RCC | NCT02420821 | 3 | Completed | Combination with atezolizumab | [53] | ||
| Vandetanib (ZD6474) | VEGFR | Metastatic Papillary or Follicular Thyroid Cancer | NCT00537095 | 2 | Active, not recruiting | Tested for patients with thyroid neoplasms which are failing or unsuitable for radioiodine therapy | [54] |
| Axitinib (AG-013738) | VEGFR | Metastatic RCC | NCT00920816 | 3 | Completed | Tested as single agent compared to sorafenib | [55] |
| Siltuximab | IL6 | Metastatic Pancreatic Cancer | NCT04191421 | 1–2 | Recruiting | Combination with spartalizumab | |
| Tocilizumab | IL-R6 | Melanoma; NSCLC | NCT04940299 | 2 | Recruiting | Combination with ipilimumab and nivolumab | [56] |
2.1.2. Toll-Like Receptors (TLRs)
2.1.3. Cellular Inhibitor of Apoptosis Proteins (c-IAPs)
2.1.4. The Phosphoinositide 3-Kinase (PI3K)/AKT Pathway
2.1.5. B Cell Receptor (BCR) Signaling
2.2. Agents Targeting Core Components of NF-κB Pathway
2.2.1. IKK Complex
2.2.2. Ubiquitin and Proteasome Pathway
2.2.3. NF-κB Transcription Factors
2.2.4. NF-κB Nuclear Activities
2.3. Inhibitors of NF-κB Downstream Effectors
3. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Multiple Nuclear Factors Interact with the Immunoglobulin Enhancer Sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-Kappa B System: A Treasure Trove for Drug Development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-KappaB Signaling: 785 and Counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-ΚB, the First Quarter-Century: Remarkable Progress and Outstanding Questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-ΚB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Staudt, L.M. Oncogenic Activation of NF-KappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The Complexity of NF-ΚB Signaling in Inflammation and Cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Hinz, M.; Scheidereit, C. The IκB Kinase Complex in NF-ΚB Regulation and Beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef]
- Cildir, G.; Low, K.C.; Tergaonkar, V. Noncanonical NF-ΚB Signaling in Health and Disease. Trends Mol. Med. 2016, 22, 414–429. [Google Scholar] [CrossRef]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-ΚB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D.; Gilmore, T.D. Good Cop, Bad Cop: The Different Faces of NF-KappaB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-Ubiquitin Ligase Complex Associates Specifically with Phosphorylated Destruction Motifs in IkappaBalpha and Beta-Catenin and Stimulates IkappaBalpha Ubiquitination in Vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.E.; Mitxitorena, I.; Carmody, R.J. The Ubiquitination of NF-ΚB Subunits in the Control of Transcription. Cells 2016, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-KappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Bonizzi, G.; Karin, M. The Two NF-KappaB Activation Pathways and Their Role in Innate and Adaptive Immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A Single NFκB System for Both Canonical and Non-Canonical Signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Annemann, M.; Plaza-Sirvent, C.; Schmitz, I. Atypical IκB Proteins-Nuclear Modulators of NF-ΚB Signaling. Cell Commun. Signal. 2013, 11, 23. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Capece, D.; Verzella, D.; Tessitore, A.; Alesse, E.; Capalbo, C.; Zazzeroni, F. Cancer Secretome and Inflammation: The Bright and the Dark Sides of NF-ΚB. Semin. Cell Dev. Biol. 2018, 78, 51–61. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-ΚB and the Link between Inflammation and Cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse Large B-Cell Lymphoma-Treatment Approaches in the Molecular Era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.E.; Morshed, R.A.; Yamini, B. Nuclear Factor-ΚB in Glioblastoma: Insights into Regulators and Targeted Therapy. Neuro. Oncol. 2016, 18, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, Y.N.; Glebov, O.K.; Zingone, A.; Keats, J.J.; Bergsagel, P.L.; Kuehl, W.M. Classical and/or Alternative NF-KappaB Pathway Activation in Multiple Myeloma. Blood 2010, 115, 3541–3552. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-Receptors: From Mediators of Cell Death and Inflammation to Therapeutic Giants-Past, Present and Future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef]
- Gerriets, V.; Goyal, A.; Khaddour, K. Tumor Necrosis Factor Inhibitors. In Treasure Island (FL); StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Jakob, J.; Hohenberger, P. Role of Isolated Limb Perfusion with Recombinant Human Tumor Necrosis Factor α and Melphalan in Locally Advanced Extremity Soft Tissue Sarcoma. Cancer 2016, 122, 2624–2632. [Google Scholar] [CrossRef]
- O’Connell, J.; Porter, J.; Kroeplien, B.; Norman, T.; Rapecki, S.; Davis, R.; McMillan, D.; Arakaki, T.; Burgin, A.; Fox Iii, D.; et al. Small Molecules That Inhibit TNF Signalling by Stabilising an Asymmetric Form of the Trimer. Nat. Commun. 2019, 10, 5795. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Cronin, S.J.F.; Penninger, J.M. Targeting the RANKL/RANK/OPG Axis for Cancer Therapy. Front. Oncol. 2020, 10, 1283. [Google Scholar] [CrossRef]
- Hildebrand, G.K.; Kasi, A. Denosumab. In Treasure Island (FL); StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Jiang, M.; Peng, L.; Yang, K.; Wang, T.; Yan, X.; Jiang, T.; Xu, J.; Qi, J.; Zhou, H.; Qian, N.; et al. Development of Small-Molecules Targeting Receptor Activator of Nuclear Factor-ΚB Ligand (RANKL)-Receptor Activator of Nuclear Factor-ΚB (RANK) Protein-Protein Interaction by Structure-Based Virtual Screening and Hit Optimization. J. Med. Chem. 2019, 62, 5370–5381. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Lue, J.K.; Sawas, A.; Amengual, J.E.; Deng, C.; Kalac, M.; Falchi, L.; Marchi, E.; Turenne, I.; Lichtenstein, R.; et al. Brentuximab Vedotin plus Bendamustine in Relapsed or Refractory Hodgkin’s Lymphoma: An International, Multicentre, Single-Arm, Phase 1-2 Trial. Lancet Oncol. 2018, 19, 257–266. [Google Scholar] [CrossRef]
- Gotlib, J.; Baird, J.H.; George, T.I.; Langford, C.; Reyes, I.; Abuel, J.; Perkins, C.; Schroeder, K.; Bose, P.; Verstovsek, S. A Phase 2 Study of Brentuximab Vedotin in Patients with CD30-Positive Advanced Systemic Mastocytosis. Blood Adv. 2019, 3, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Guo, Y.; Huang, H.; Li, W.; Ke, X.; Feng, J.; Xu, W.; Miao, H.; Kinley, J.; Song, G.; et al. Phase II Single-Arm Study of Brentuximab Vedotin in Chinese Patients with Relapsed/Refractory Classical Hodgkin Lymphoma or Systemic Anaplastic Large Cell Lymphoma. Expert Rev. Hematol. 2021, 14, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Fanale, M.A.; Moskowitz, C.H.; Shustov, A.R.; Mitra, S.; Ye, W.; Younes, A.; Moskowitz, A.J. Phase II Study of Idelalisib, a Selective Inhibitor of PI3Kδ, for Relapsed/Refractory Classical Hodgkin Lymphoma. Ann. Oncol. 2017, 28, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Novo, M.; Castellino, A.; Chiappella, A.; Ciccone, G.; Balzarotti, M.; Di Rocco, A.; Spina, M.; Vitolo, U. Copanlisib in Combination with Rituximab-Bendamustine in Patients with Relapsed-Refractory Dlbcl: A Multicentric Phase II Trial of the Fondazione Italiana Linfomi. Hematol. Oncol. 2021, 39. [Google Scholar] [CrossRef]
- Grunenberg, A.; Kaiser, L.M.; Woelfle, S.; Schmelzle, B.; Viardot, A.; Möller, P.; Barth, T.F.E.; Muche, R.; Dreyhaupt, J.; Raderer, M.; et al. A Phase II Study of the PI3K Inhibitor Copanlisib in Combination with the Anti-CD20 Monoclonal Antibody Rituximab for Patients with Marginal Zone Lymphoma: Treatment Rationale and Protocol Design of the COUP-1 Trial. BMC Cancer 2021, 21, 749. [Google Scholar] [CrossRef]
- Chen, L.S.; Bose, P.; Cruz, N.D.; Jiang, Y.; Wu, Q.; Thompson, P.A.; Feng, S.; Kroll, M.H.; Qiao, W.; Huang, X.; et al. A Pilot Study of Lower Doses of Ibrutinib in Patients with Chronic Lymphocytic Leukemia. Blood 2018, 132, 2249–2259. [Google Scholar] [CrossRef]
- Le Gouill, S.; Morschhauser, F.; Chiron, D.; Bouabdallah, K.; Cartron, G.; Casasnovas, O.; Bodet-Milin, C.; Ragot, S.; Bossard, C.; Nadal, N.; et al. Ibrutinib, Obinutuzumab, and Venetoclax in Relapsed and Untreated Patients with Mantle Cell Lymphoma: A Phase 1/2 Trial. Blood 2021, 137, 877–887. [Google Scholar] [CrossRef]
- Jain, N.; Ferrajoli, A.; Basu, S.; Thompson, P.A.; Burger, J.A.; Kadia, T.M.; Estrov, Z.E.; Pemmaraju, N.; Lopez, W.; Thakral, B.; et al. A Phase II Trial of Nivolumab Combined with Ibrutinib for Patients with Richter Transformation. Blood 2018, 132, 296. [Google Scholar] [CrossRef]
- Mo, D.; Zhu, H.; Wang, J.; Hao, H.; Guo, Y.; Wang, J.; Han, X.; Zou, L.; Li, Z.; Yao, H.; et al. Icaritin Inhibits PD-L1 Expression by Targeting Protein IκB Kinase α. Eur. J. Immunol. 2021, 51, 978–988. [Google Scholar] [CrossRef]
- Quillin, J.; Patel, R.; Herzberg, E.; Alton, D.; Bikzhanova, G.; Geisler, L.; Olson, J. A Phase 0 Analysis of Ixazomib in Patients with Glioblastoma. Mol. Clin. Oncol. 2020, 13, 43. [Google Scholar] [CrossRef]
- Zhou, X.; Sedarati, F.; Faller, D.V.; Zhao, D.; Faessel, H.M.; Chowdhury, S.; Bolleddula, J.; Li, Y.; Venkatakrishnan, K.; Papai, Z. Phase I Study Assessing the Mass Balance, Pharmacokinetics, and Excretion of [(14)C]-Pevonedistat, a NEDD8-Activating Enzyme Inhibitor in Patients with Advanced Solid Tumors. Investig. New Drugs 2021, 39, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-Week Selinexor, Bortezomib, and Dexamethasone versus Twice-per-Week Bortezomib and Dexamethasone in Patients with Multiple Myeloma (BOSTON): A Randomised, Open-Label, Phase 3 Trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Castillo, J.J.; Allan, J.N.; Siddiqi, T.; Advani, R.H.; Meid, K.; Leventoff, C.; White, T.P.; Flynn, C.A.; Sarosiek, S.; Branagan, A.R.; et al. Venetoclax in Previously Treated Waldenström Macroglobulinemia. J. Clin. Oncol. 2022, 40, 63–71. [Google Scholar] [CrossRef]
- Davis, J.E.; Handunnetti, S.M.; Ludford-Menting, M.; Sharpe, C.; Blombery, P.; Anderson, M.A.; Roberts, A.W.; Seymour, J.F.; Tam, C.S.; Ritchie, D.S.; et al. Immune Recovery in Patients with Mantle Cell Lymphoma Receiving Long-Term Ibrutinib and Venetoclax Combination Therapy. Blood Adv. 2020, 4, 4849–4859. [Google Scholar] [CrossRef]
- Pollyea, D.A.; DiNardo, C.D.; Arellano, M.L.; Pigneux, A.; Fiedler, W.; Konopleva, M.; Rizzieri, D.A.; Smith, B.D.; Shinagawa, A.; Lemoli, R.M.; et al. Impact of Venetoclax and Azacitidine in Treatment-Naïve Patients with Acute Myeloid Leukemia and IDH1/2 Mutations. Clin. Cancer Res. 2022, 28, 2753–2761. [Google Scholar] [CrossRef]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.; Greil, R.; Illés, Á.; Johnson, N.A.; Larouche, J.-F.; Lugtenburg, P.J.; Patti, C.; et al. A Phase 2 Study of Venetoclax plus R-CHOP as First-Line Treatment for Patients with Diffuse Large B-Cell Lymphoma. Blood 2021, 137, 600–609. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Salles, G.; Mason, K.D.; Casulo, C.; Le Gouill, S.; Sehn, L.H.; Tilly, H.; Cartron, G.; Chamuleau, M.E.D.; Goy, A.; et al. Venetoclax plus R- or G-CHOP in Non-Hodgkin Lymphoma: Results from the CAVALLI Phase 1b Trial. Blood 2019, 133, 1964–1976. [Google Scholar] [CrossRef]
- Rogers, K.A.; Huang, Y.; Ruppert, A.S.; Abruzzo, L.V.; Andersen, B.L.; Awan, F.T.; Bhat, S.A.; Dean, A.; Lucas, M.; Banks, C.; et al. Phase II Study of Combination Obinutuzumab, Ibrutinib, and Venetoclax in Treatment-Naïve and Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2020, 38, 3626–3637. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Rini, B.I.; Motzer, R.J.; Powles, T.; McDermott, D.F.; Escudier, B.; Donskov, F.; Hawkins, R.; Bracarda, S.; Bedke, J.; De Giorgi, U.; et al. Atezolizumab plus Bevacizumab Versus Sunitinib for Patients with Untreated Metastatic Renal Cell Carcinoma and Sarcomatoid Features: A Prespecified Subgroup Analysis of the IMmotion151 Clinical Trial. Eur. Urol. 2021, 79, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Bastholt, L.; Krause, T.; de la Fouchardiere, C.; Tennvall, J.; Awada, A.; Gómez, J.M.; Bonichon, F.; Leenhardt, L.; Soufflet, C.; et al. Vandetanib in Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 2 Trial. Lancet Oncol. 2012, 13, 897–905. [Google Scholar] [CrossRef]
- Hutson, T.E.; Al-Shukri, S.; Stus, V.P.; Lipatov, O.N.; Shparyk, Y.; Bair, A.H.; Rosbrook, B.; Andrews, G.I.; Vogelzang, N.J. Axitinib Versus Sorafenib in First-Line Metastatic Renal Cell Carcinoma: Overall Survival From a Randomized Phase III Trial. Clin. Genitourin. Cancer 2017, 15, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, N.; Montazari, E.; Spillson, C.; Bentebibel, S.-E.; Awiwi, M.; Elsayes, K.M.; Gao, J.; Altan, M.; Wong, M.K.K.; Glitza, I.C.; et al. Tocilizumab in Combination with Ipilimumab and Nivolumab in Solid Tumors. J. Clin. Oncol. 2022, 40, TPS9600. [Google Scholar] [CrossRef]
- Wang, J.Q.; Jeelall, Y.S.; Ferguson, L.L.; Horikawa, K. Toll-Like Receptors and Cancer: MYD88 Mutation and Inflammation. Front. Immunol. 2014, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Idera FDA Orphan Drug Designation for IMO-8400 for the Treatment of Diffuse Large b-Cell Lymphoma. Available online: https://ir.iderapharma.com/news-releases/news-release-details/idera-announces-fda-orphan-drug-designation-imo-8400-treatment (accessed on 31 July 2022).
- Idera Update on IMO-9200, an Antagonist of Toll-like Receptors. Available online: https://ir.iderapharma.com/news-releases/news-release-details/idera-pharmaceuticals-provides-development-update-imo-9200 (accessed on 31 July 2022).
- Hajam, I.A.; Dar, P.A.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial Flagellin-a Potent Immunomodulatory Agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef]
- Liu, G.; Park, Y.-J.; Abraham, E. Interleukin-1 Receptor-Associated Kinase (IRAK) -1- Mediated NF-κ Activation Requires Cytosolic and Nuclear Activity. FASEB J. 2008, 22, 2285–2296. [Google Scholar] [CrossRef]
- Suzuki, N.; Saito, T. IRAK-4–a Shared NF-ΚB Activator in Innate and Acquired Immunity. Trends Immunol. 2006, 27, 566–572. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK Signalling in Cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef]
- Srivastava, R.; Geng, D.; Liu, Y.; Zheng, L.; Li, Z.; Joseph, M.A.; McKenna, C.; Bansal, N.; Ochoa, A.; Davila, E. Augmentation of Therapeutic Responses in Melanoma by Inhibition of IRAK-1,-4. Cancer Res. 2012, 72, 6209–6216. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. IAPs: From Caspase Inhibitors to Modulators of NF-KappaB, Inflammation and Cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, C.A.; Mitsuuchi, Y.; Burns, J.M.; Neiman, E.M.; Condon, S.M.; Yu, G.; Seipel, M.E.; Kapoor, G.S.; Laporte, M.G.; Rippin, S.R.; et al. Birinapant (TL32711), a Bivalent SMAC Mimetic, Targets TRAF2-Associated CIAPs, Abrogates TNF-Induced NF-ΚB Activation, and Is Active in Patient-Derived Xenograft Models. Mol. Cancer Ther. 2014, 13, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Tao, Y.; Gomez-Roca, C.; Cristina, V.; Borcoman, E.; Deutsch, E.; Bahleda, R.; Calugaru, V.; Modesto, A.; Rouits, E.; et al. Phase I Trial of Debio 1143, an Antagonist of Inhibitor of Apoptosis Proteins, Combined with Cisplatin Chemoradiotherapy in Patients with Locally Advanced Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2020, 26, 6429–6436. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-S.; Tao, Y.; Le Tourneau, C.; Pointreau, Y.; Sire, C.; Kaminsky, M.-C.; Coutte, A.; Alfonsi, M.; Boisselier, P.; Martin, L.; et al. Debio 1143 and High-Dose Cisplatin Chemoradiotherapy in High-Risk Locoregionally Advanced Squamous Cell Carcinoma of the Head and Neck: A Double-Blind, Multicentre, Randomised, Phase 2 Study. Lancet Oncol. 2020, 21, 1173–1187. [Google Scholar] [CrossRef]
- Debio 1143. Available online: https://www.debiopharm.com/drug-development/press-releases/fda-grants-orphan-drug-designation-to-debiopharm-international-sas-iap-inhibitor-debio-1143-in-the-treatment-of-ovarian-cancer/ (accessed on 31 July 2022).
- Pan, W.; Luo, Q.; Yan, X.; Yuan, L.; Yi, H.; Zhang, L.; Li, B.; Zhang, Y.; Sun, J.; Qiu, M.-Z.; et al. A Novel SMAC Mimetic APG-1387 Exhibits Dual Antitumor Effect on HBV-Positive Hepatocellular Carcinoma with High Expression of CIAP2 by Inducing Apoptosis and Enhancing Innate Anti-Tumor Immunity. Biochem. Pharmacol. 2018, 154, 127–135. [Google Scholar] [CrossRef]
- Li, B.-X.; Wang, H.-B.; Qiu, M.-Z.; Luo, Q.-Y.; Yi, H.-J.; Yan, X.-L.; Pan, W.-T.; Yuan, L.-P.; Zhang, Y.-X.; Xu, J.-H.; et al. Correction to: Novel Smac Mimetic APG-1387 Elicits Ovarian Cancer Cell Killing through TNF-Alpha, Ripoptosome and Autophagy Mediated Cell Death Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 108. [Google Scholar] [CrossRef]
- Li, N.; Feng, L.; Han, H.-Q.; Yuan, J.; Qi, X.-K.; Lian, Y.-F.; Kuang, B.-H.; Zhang, Y.-C.; Deng, C.-C.; Zhang, H.-J.; et al. A Novel Smac Mimetic APG-1387 Demonstrates Potent Antitumor Activity in Nasopharyngeal Carcinoma Cells by Inducing Apoptosis. Cancer Lett. 2016, 381, 14–22. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Cheung, C.H.A. An Updated Review of Smac Mimetics, LCL161, Birinapant, and GDC-0152 in Cancer Treatment. Appl. Sci. 2021, 11, 335. [Google Scholar] [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT Pathway in Cancer: The Framework of Malignant Behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ji, M. Receptor Tyrosine Kinases in PI3K Signaling: The Therapeutic Targets in Cancer. Semin. Cancer Biol. 2019, 59, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Mangaonkar, A. Idelalisib: A Novel PI3Kδ Inhibitor for Chronic Lymphocytic Leukemia. Ann. Pharmacother. 2015, 49, 1162–1170. [Google Scholar] [CrossRef]
- Zirlik, K.; Veelken, H. Idelalisib. Recent Results Cancer Res. 2018, 212, 243–264. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Follows, G.A.; Nastoupil, L.J. Copanlisib for the Treatment of Malignant Lymphoma: Clinical Experience and Future Perspectives. Target. Oncol. 2021, 16, 295–308. [Google Scholar] [CrossRef]
- Alpelisib. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-alpelisib-metastatic-breast-cancer (accessed on 31 July 2022).
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Phosphoinositide 3-Kinases. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef]
- Wright, S.C.E.; Vasilevski, N.; Serra, V.; Rodon, J.; Eichhorn, P.J.A. Mechanisms of Resistance to PI3K Inhibitors in Cancer: Adaptive Responses, Drug Tolerance and Cellular Plasticity. Cancers 2021, 13, 1538. [Google Scholar] [CrossRef]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef] [PubMed]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and Consequences of Constitutive NF-ΚB Activation in B-Cell Lymphoid Malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Phelan, J.D.; Wilson, W.H.; Staudt, L.M. Pathogenic B-Cell Receptor Signaling in Lymphoid Malignancies: New Insights to Improve Treatment. Immunol. Rev. 2019, 291, 190–213. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.; Klein, U. Aberrant Activation of NF-ΚB Signalling in Aggressive Lymphoid Malignancies. Cells 2018, 7, 189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Smith, S.M.; Zhang, S.Y.; Lynn Wang, Y. Mechanisms of Ibrutinib Resistance in Chronic Lymphocytic Leukaemia and Non-Hodgkin Lymphoma. Br. J. Haematol. 2015, 170, 445–456. [Google Scholar] [CrossRef]
- FDA. Available online: https://www.fda.gov/ (accessed on 31 July 2022).
- Wilson, W.H.; Wright, G.W.; Huang, D.W.; Hodkinson, B.; Balasubramanian, S.; Fan, Y.; Vermeulen, J.; Shreeve, M.; Staudt, L.M. Effect of Ibrutinib with R-CHOP Chemotherapy in Genetic Subtypes of DLBCL. Cancer Cell 2021, 39, 1643–1653.e3. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, N.L.; Costello, B.A.; LaPlant, B.R.; Ansell, S.M.; Kuruvilla, J.G.; Reeder, C.B.; Thye, L.S.; Anderson, D.M.; Krysiak, K.; Ramirez, C.; et al. Single-Agent Ibrutinib in Relapsed or Refractory Follicular Lymphoma: A Phase 2 Consortium Trial. Blood 2018, 131, 182–190. [Google Scholar] [CrossRef]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L., III; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Antonia, R.J.; Hagan, R.S.; Baldwin, A.S. Expanding the View of IKK: New Substrates and New Biology. Trends Cell Biol. 2021, 31, 166–178. [Google Scholar] [CrossRef]
- Karin, M. Whipping NF-ΚB to Submission via GADD45 and MKK7. Cancer Cell 2014, 26, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory Kappa B Kinases as Targets for Pharmacological Regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Grothe, K.; Flechsenhar, K.; Paehler, T.; Ritzeler, O.; Beninga, J.; Saas, J.; Herrmann, M.; Rudolphi, K. IκB Kinase Inhibition as a Potential Treatment of Osteoarthritis-Results of a Clinical Proof-of-Concept Study. Osteoarthr. Cartil. 2017, 25, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-ΚB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Hovstadius, P.; Larsson, R.; Jonsson, E.; Skov, T.; Kissmeyer, A.-M.; Krasilnikoff, K.; Bergh, J.; Karlsson, M.O.; Lönnebo, A.; Ahlgren, J. A Phase I Study of CHS 828 in Patients with Solid Tumor Malignancy. Clin. Cancer Res. 2002, 8, 2843–2850. [Google Scholar]
- Xu, G.; Lo, Y.-C.; Li, Q.; Napolitano, G.; Wu, X.; Jiang, X.; Dreano, M.; Karin, M.; Wu, H. Crystal Structure of Inhibitor of ΚB Kinase β. Nature 2011, 472, 325–330. [Google Scholar] [CrossRef]
- Ullah, M.A.; Johora, F.T.; Sarkar, B.; Araf, Y.; Rahman, M.H. Curcumin Analogs as the Inhibitors of TLR4 Pathway in Inflammation and Their Drug like Potentialities: A Computer-Based Study. J. Recept. Signal Transduct. Res. 2020, 40, 324–338. [Google Scholar] [CrossRef]
- Cho, Y.W.; Lim, H.J.; Han, M.H.; Kim, B.-C.; Han, S. Small Molecule Inhibitors of IκB Kinase β: A Chip-Based Screening and Molecular Docking Simulation. Bioorg. Med. Chem. 2020, 28, 115440. [Google Scholar] [CrossRef]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glöckner, J.; Pober, J.S.; Ghosh, S. Selective Inhibition of NF-KappaB Activation by a Peptide That Blocks the Interaction of NEMO with the IkappaB Kinase Complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Gao, J.; Zhang, X.; Peng, Y.; Wei, W.; Xu, J.; Li, Z.; Wang, C.; Zhou, M.; Tian, X.; et al. Characterization of a Small-Molecule Inhibitor Targeting NEMO/IKKβ to Suppress Colorectal Cancer Growth. Signal Transduct. Target. Ther. 2022, 7, 71. [Google Scholar] [CrossRef]
- Llona-Minguez, S.; Baiget, J.; Mackay, S.P. Small-Molecule Inhibitors of IκB Kinase (IKK) and IKK-Related Kinases. Pharm. Pat. Anal. 2013, 2, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, V.V.; Mathison, J.C.; Schwamborn, K.; Mercurio, F.; Ulevitch, R.J. IKKi/IKKepsilon Plays a Key Role in Integrating Signals Induced by pro-Inflammatory Stimuli. J. Biol. Chem. 2003, 278, 26612–26619. [Google Scholar] [CrossRef] [PubMed]
- Adli, M.; Baldwin, A.S. IKK-i/IKKepsilon Controls Constitutive, Cancer Cell-Associated NF-KappaB Activity via Regulation of Ser-536 P65/RelA Phosphorylation. J. Biol. Chem. 2006, 281, 26976–26984. [Google Scholar] [CrossRef]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small Molecules Targeting the Innate Immune CGAS–STING–TBK1 Signaling Pathway. Acta Pharm. Sin. B 2020, 10, 2272–2298. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Yan, N. Therapeutic Potential of Targeting TBK1 in Autoimmune Diseases and Interferonopathies. Pharmacol. Res. 2016, 111, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M. IKKepsilon: A Bridge between Obesity and Inflammation. Cell 2009, 138, 834–836. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Welsh, E.A.; Oguz, U.; Fang, B.; Bai, Y.; Kinose, F.; Bronk, C.; Remsing Rix, L.L.; Beg, A.A.; Rix, U.; et al. Dissection of TBK1 Signaling via Phosphoproteomics in Lung Cancer Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12414–12419. [Google Scholar] [CrossRef]
- Barbie, T.U.; Alexe, G.; Aref, A.R.; Li, S.; Zhu, Z.; Zhang, X.; Imamura, Y.; Thai, T.C.; Huang, Y.; Bowden, M.; et al. Targeting an IKBKE Cytokine Network Impairs Triple-Negative Breast Cancer Growth. J. Clin. Investig. 2014, 124, 5411–5423. [Google Scholar] [CrossRef]
- Thomson, D.W.; Bergamini, G. Recent Progress in Small Molecule TBK1 Inhibitors: A Patent Review (2015-2020). Expert Opin. Ther. Pat. 2021, 31, 785–794. [Google Scholar] [CrossRef]
- Thomson, D.W.; Poeckel, D.; Zinn, N.; Rau, C.; Strohmer, K.; Wagner, A.J.; Graves, A.P.; Perrin, J.; Bantscheff, M.; Duempelfeld, B.; et al. Discovery of GSK8612, a Highly Selective and Potent TBK1 Inhibitor. ACS Med. Chem. Lett. 2019, 10, 780–785. [Google Scholar] [CrossRef]
- Reilly, S.M.; Chiang, S.-H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An Inhibitor of the Protein Kinases TBK1 and IKK-ɛ Improves Obesity-Related Metabolic Dysfunctions in Mice. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyett, T.S.; Gan, X.; Reilly, S.M.; Chang, L.; Gomez, A.V.; Saltiel, A.R.; Showalter, H.D.; Tesmer, J.J.G. Carboxylic Acid Derivatives of Amlexanox Display Enhanced Potency toward TBK1 and IKKε and Reveal Mechanisms for Selective Inhibition. Mol. Pharmacol. 2018, 94, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Beyett, T.S.; Gan, X.; Reilly, S.M.; Gomez, A.V.; Chang, L.; Tesmer, J.J.G.; Saltiel, A.R.; Showalter, H.D. Design, Synthesis, and Biological Activity of Substituted 2-Amino-5-Oxo-5H-Chromeno[2,3-b]Pyridine-3-Carboxylic Acid Derivatives as Inhibitors of the Inflammatory Kinases TBK1 and IKKε for the Treatment of Obesity. Bioorg. Med. Chem. 2018, 26, 5443–5461. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, H.; Kaniskan, H.Ü.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef]
- Arvinas PROTAC Targeted Protein Degradation. Available online: https://www.arvinas.com/ (accessed on 31 July 2022).
- Cheng, J.; Feng, X.; Li, Z.; Zhou, F.; Yang, J.-M.; Zhao, Y. Pharmacological Inhibition of NF-ΚB-Inducing Kinase (NIK) with Small Molecules for the Treatment of Human Diseases. RSC Med. Chem. 2021, 12, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Mortier, J.; Masereel, B.; Remouchamps, C.; Ganeff, C.; Piette, J.; Frederick, R. NF-KappaB Inducing Kinase (NIK) Inhibitors: Identification of New Scaffolds Using Virtual Screening. Bioorg. Med. Chem. Lett. 2010, 20, 4515–4520. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Su, M.-B.; Gao, L.-X.; Zhou, Y.-B.; Yuan, B.; Lyu, X.; Yan, Z.; Hu, C.; Zhang, H.; et al. Discovery of a Potent and Selective NF-ΚB-Inducing Kinase (NIK) Inhibitor That Has Anti-Inflammatory Effects in Vitro and in Vivo. J. Med. Chem. 2020, 63, 4388–4407. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, X.; Jia, L.; Chen, D.; Hou, H.; Rui, L.; Zhao, Y.; Chen, Z. A Small-Molecule Inhibitor of NF-ΚB-Inducing Kinase (NIK) Protects Liver from Toxin-Induced Inflammation, Oxidative Stress, and Injury. FASEB J. 2017, 31, 711–718. [Google Scholar] [CrossRef]
- Hu, C.; Xia, H.; Bai, S.; Zhao, J.; Edwards, H.; Li, X.; Yang, Y.; Lyu, J.; Wang, G.; Zhan, Y.; et al. CUDC-907, a Novel Dual PI3K and HDAC Inhibitor, in Prostate Cancer: Antitumour Activity and Molecular Mechanism of Action. J. Cell. Mol. Med. 2020, 24, 7239–7253. [Google Scholar] [CrossRef]
- Brightbill, H.D.; Suto, E.; Blaquiere, N.; Ramamoorthi, N.; Sujatha-Bhaskar, S.; Gogol, E.B.; Castanedo, G.M.; Jackson, B.T.; Kwon, Y.C.; Haller, S.; et al. NF-ΚB Inducing Kinase Is a Therapeutic Target for Systemic Lupus Erythematosus. Nat. Commun. 2018, 9, 179. [Google Scholar] [CrossRef]
- Versele, M.; Janssen, L.; Geerts, T.; Floren, W.; Janssens, B.; Millar, H.; Jacoby, E.; Gross, G.; Ligny, Y.; Simonnet, Y.; et al. Abstract 4199: Inhibition of NF-KB Inducing Kinase (NIK) Selectively Abrogates NIK and TRAF3 Mutant Multiple Myeloma Tumor Growth. Cancer Res. 2017, 77, 4199. [Google Scholar] [CrossRef]
- LaPlante, G.; Zhang, W. Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers 2021, 13, 3079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin-Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Kolibaba, K.S.; Reeves, J.A.; Tulpule, A.; Flinn, I.W.; Kolevska, T.; Robles, R.; Flowers, C.R.; Collins, R.; DiBella, N.J.; et al. Randomized Phase II Study of R-CHOP With or Without Bortezomib in Previously Untreated Patients With Non-Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017, 35, 3538–3546. [Google Scholar] [CrossRef]
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-Expression Profiling of Bortezomib Added to Standard Chemoimmunotherapy for Diffuse Large B-Cell Lymphoma (REMoDL-B): An Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2019, 20, 649–662. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, Bortezomib, and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma Previously Treated with Lenalidomide (OPTIMISMM): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef]
- Luttwak, E.; Gatt, M.E.; Lebel, E.; Lavi, N.; Tadmor, T.; Natalia, K.; Benyamini, N.; Horowitz, N.; Geva, M.; Suriu, C.; et al. Bortezomib Maintenance Therapy as a Standard of Care Provides Favorable Outcomes in Newly Diagnosed Myeloma Patients: A Multisite Real-Life Study. Clin. Lymphoma. Myeloma Leuk. 2020, 20, e850–e857. [Google Scholar] [CrossRef]
- Sivaraj, D.; Green, M.M.; Li, Z.; Sung, A.D.; Sarantopoulos, S.; Kang, Y.; Long, G.D.; Horwitz, M.E.; Lopez, R.D.; Sullivan, K.M.; et al. Outcomes of Maintenance Therapy with Bortezomib after Autologous Stem Cell Transplantation for Patients with Multiple Myeloma. Biol. Blood Marrow Transplant. 2017, 23, 262–268. [Google Scholar] [CrossRef]
- Suzuki, E.; Demo, S.; Deu, E.; Keats, J.; Arastu-Kapur, S.; Bergsagel, P.L.; Bennett, M.K.; Kirk, C.J. Molecular Mechanisms of Bortezomib Resistant Adenocarcinoma Cells. PLoS ONE 2011, 6, e27996. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of Therapy with Bortezomib in Solid Tumors: A Review Based on 32 Clinical Trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Perel, G.; Bliss, J.; Thomas, C.M. Carfilzomib (Kyprolis): A Novel Proteasome Inhibitor for Relapsed And/or Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 303–307. [Google Scholar]
- Usmani, S.Z.; Quach, H.; Mateos, M.-V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Gavriatopoulou, M.; Oriol, A.; Rabin, N.; et al. Carfilzomib, Dexamethasone, and Daratumumab versus Carfilzomib and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma (CANDOR): Updated Outcomes from a Randomised, Multicentre, Open-Label, Phase 3 Study. Lancet Oncol. 2022, 23, 65–76. [Google Scholar] [CrossRef]
- Hari, P.; Paba-Prada, C.E.; Voorhees, P.M.; Frye, J.; Chang, Y.-L.; Moreau, P.; Zonder, J.; Boccia, R.; Shain, K.H. Efficacy and Safety Results from a Phase 1b/2, Multicenter, Open-Label Study of Oprozomib and Dexamethasone in Patients with Relapsed and/or Refractory Multiple Myeloma. Leuk. Res. 2019, 83, 106172. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Usmani, S.; Stadtmauer, E.A.; Rifkin, R.M.; Berenson, J.R.; Berdeja, J.G.; Lyons, R.M.; Klippel, Z.; Chang, Y.-L.; Niesvizky, R. Oprozomib, Pomalidomide, and Dexamethasone in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma. Myeloma Leuk. 2019, 19, 570–578.e1. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Vij, R.; Siegel, D.; Badros, A.; Kaufman, J.; Raje, N.; Jakubowiak, A.; Savona, M.R.; Obreja, M.; Berdeja, J.G. A Phase Ib/II Study of Oprozomib in Patients with Advanced Multiple Myeloma and Waldenström Macroglobulinemia. Clin. Cancer Res. 2019, 25, 4907–4916. [Google Scholar] [CrossRef]
- Hari, P.; Matous, J.V.; Voorhees, P.M.; Shain, K.H.; Obreja, M.; Frye, J.; Fujii, H.; Jakubowiak, A.J.; Rossi, D.; Sonneveld, P. Oprozomib in Patients with Newly Diagnosed Multiple Myeloma. Blood Cancer J. 2019, 9, 66. [Google Scholar] [CrossRef]
- Gupta, N.; Hanley, M.J.; Xia, C.; Labotka, R.; Harvey, R.D.; Venkatakrishnan, K. Clinical Pharmacology of Ixazomib: The First Oral Proteasome Inhibitor. Clin. Pharmacokinet. 2019, 58, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Poenisch, W.; Knop, S.; Egle, A.; Schreder, M.; Lechner, D.; Hajek, R.; Gunsilius, E.; Krenosz, K.J.; Petzer, A.; et al. Ixazomib-Thalidomide-Dexamethasone for Induction Therapy Followed by Ixazomib Maintenance Treatment in Patients with Relapsed/Refractory Multiple Myeloma. Br. J. Cancer 2019, 121, 751–757. [Google Scholar] [CrossRef]
- Auner, H.W.; Brown, S.R.; Walker, K.; Kendall, J.; Dawkins, B.; Meads, D.; Morgan, G.J.; Kaiser, M.F.; Cook, M.; Roberts, S.; et al. Ixazomib with Cyclophosphamide and Dexamethasone in Relapsed or Refractory Myeloma: MUKeight Phase II Randomised Controlled Trial Results. Blood Cancer J. 2022, 12, 52. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Špička, I.; Quach, H.; Oriol, A.; Hájek, R.; Garg, M.; Beksac, M.; Bringhen, S.; Katodritou, E.; Chng, W.-J.; et al. Ixazomib as Postinduction Maintenance for Patients With Newly Diagnosed Multiple Myeloma Not Undergoing Autologous Stem Cell Transplantation: The Phase III TOURMALINE-MM4 Trial. J. Clin. Oncol. 2020, 38, 4030–4041. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Kastritis, E.; Wechalekar, A.D.; Schönland, S.O.; Kim, K.; Sanchorawala, V.; Landau, H.J.; Kwok, F.; Suzuki, K.; Comenzo, R.L.; et al. A Randomized Phase 3 Study of Ixazomib-Dexamethasone versus Physician’s Choice in Relapsed or Refractory AL Amyloidosis. Leukemia 2022, 36, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.J.; Fenical, W.; et al. Marizomib, a Proteasome Inhibitor for All Seasons: Preclinical Profile and a Framework for Clinical Trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Zimmerman, T.M.; Hofmeister, C.C.; Talpaz, M.; Chanan-Khan, A.A.; Kaufman, J.L.; Laubach, J.P.; Chauhan, D.; Jakubowiak, A.J.; Reich, S.; et al. Phase 1 Study of Marizomib in Relapsed or Relapsed and Refractory Multiple Myeloma: NPI-0052-101 Part 1. Blood 2016, 127, 2693–2700. [Google Scholar] [CrossRef]
- Spencer, A.; Harrison, S.; Zonder, J.; Badros, A.; Laubach, J.; Bergin, K.; Khot, A.; Zimmerman, T.; Chauhan, D.; Levin, N.; et al. A Phase 1 Clinical Trial Evaluating Marizomib, Pomalidomide and Low-Dose Dexamethasone in Relapsed and Refractory Multiple Myeloma (NPI-0052-107): Final Study Results. Br. J. Haematol. 2018, 180, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef]
- Matsuo, S.; Sharma, A.; Wang, P.; Yang, W.-L. PYR-41, A Ubiquitin-Activating Enzyme E1 Inhibitor, Attenuates Lung Injury in Sepsis. Shock 2018, 49, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Meng, Y.; Wang, L.; Wang, H.-X.; Tian, C.; Pang, G.-D.; Li, H.-H.; Du, J. Ubiquitin-Activating Enzyme E1 Inhibitor PYR41 Attenuates Angiotensin II-Induced Activation of Dendritic Cells via the IκBa/NF-ΚB and MKP1/ERK/STAT1 Pathways. Immunology 2014, 142, 307–319. [Google Scholar] [CrossRef]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B.; et al. Phase Ib Study of Pevonedistat, a NEDD8-Activating Enzyme Inhibitor, in Combination with Docetaxel, Carboplatin and Paclitaxel, or Gemcitabine, in Patients with Advanced Solid Tumors. Investig. New Drugs 2019, 37, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a First-in-Class NEDD8-Activating Enzyme Inhibitor, Combined with Azacitidine in Patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef]
- Magin, R.S.; Doherty, L.M.; Buhrlage, S.J. Discovery of a First-In-Class Covalent Allosteric Inhibitor of SUMO E1 Activating Enzyme. Cell Chem. Biol. 2019, 26, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Barghout, S.H.; Patel, P.S.; Wang, X.; Xu, G.W.; Kavanagh, S.; Halgas, O.; Zarabi, S.F.; Gronda, M.; Hurren, R.; Jeyaraju, D.V.; et al. Preclinical Evaluation of the Selective Small-Molecule UBA1 Inhibitor, TAK-243, in Acute Myeloid Leukemia. Leukemia 2019, 33, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, C.; Muraoka, H.; Ochiiwa, H.; Tsuji, S.; Hashimoto, A.; Kazuno, H.; Nakagawa, F.; Komiya, Y.; Suzuki, S.; Takenaka, T.; et al. TAS4464, A Highly Potent and Selective Inhibitor of NEDD8-Activating Enzyme, Suppresses Neddylation and Shows Antitumor Activity in Diverse Cancer Models. Mol. Cancer Ther. 2019, 18, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Ochiiwa, H.; Ailiken, G.; Yokoyama, M.; Yamagata, K.; Nagano, H.; Yoshimura, C.; Muraoka, H.; Ishida, K.; Haruma, T.; Nakayama, A.; et al. TAS4464, a NEDD8-Activating Enzyme Inhibitor, Activates Both Intrinsic and Extrinsic Apoptotic Pathways via c-Myc-Mediated Regulation in Acute Myeloid Leukemia. Oncogene 2021, 40, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Shimizu, T.; Yonemori, K.; Kitano, S.; Kondo, S.; Iwasa, S.; Koyama, T.; Sudo, K.; Sato, J.; Tamura, K.; et al. A First-in-Human, Phase 1 Study of the NEDD8 Activating Enzyme E1 Inhibitor TAS4464 in Patients with Advanced Solid Tumors. Investig. New Drugs 2021, 39, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Huynh, K.Q.; Lu, I.; Moustakim, M.; Miao, H.; Yu, C.; Haeusgen, M.J.; Hopkins, B.D.; Huang, L.; Zheng, N.; et al. Inhibitors of Cullin-RING E3 Ubiquitin Ligase 4 with Antitumor Potential. Proc. Natl. Acad. Sci. USA 2021, 118, e2007328118. [Google Scholar] [CrossRef]
- Jang, C.H.; Moon, N.; Lee, J.; Kwon, M.J.; Oh, J.; Kim, J.-S. Luteolin Synergistically Enhances Antitumor Activity of Oxaliplatin in Colorectal Carcinoma via AMPK Inhibition. Antioxidants 2022, 11, 626. [Google Scholar] [CrossRef]
- Chen, L.; Ruan, Y.; Wang, X.; Min, L.; Shen, Z.; Sun, Y.; Qin, X. BAY 11-7082, a Nuclear Factor-ΚB Inhibitor, Induces Apoptosis and S Phase Arrest in Gastric Cancer Cells. J. Gastroenterol. 2014, 49, 864–874. [Google Scholar] [CrossRef]
- Kao, S.-H.; Wu, H.-T.; Wu, K.-J. Ubiquitination by HUWE1 in Tumorigenesis and Beyond. J. Biomed. Sci. 2018, 25, 67. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in Multiple Myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef]
- Crawford, L.J.; Campbell, D.C.; Morgan, J.J.; Lawson, M.A.; Down, J.M.; Chauhan, D.; McAvera, R.M.; Morris, T.C.; Hamilton, C.; Krishnan, A.; et al. The E3 Ligase HUWE1 Inhibition as a Therapeutic Strategy to Target MYC in Multiple Myeloma. Oncogene 2020, 39, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Carvajal, A.; Grishkovskaya, I.; Gomez Diaz, C.; Vogel, A.; Sonn-Segev, A.; Kushwah, M.S.; Schodl, K.; Deszcz, L.; Orban-Nemeth, Z.; Sakamoto, S.; et al. The Linear Ubiquitin Chain Assembly Complex (LUBAC) Generates Heterotypic Ubiquitin Chains. Elife 2021, 10, e60660. [Google Scholar] [CrossRef] [PubMed]
- Jo, T.; Nishikori, M.; Kogure, Y.; Arima, H.; Sasaki, K.; Sasaki, Y.; Nakagawa, T.; Iwai, F.; Momose, S.; Shiraishi, A.; et al. LUBAC Accelerates B-Cell Lymphomagenesis by Conferring Resistance to Genotoxic Stress on B Cells. Blood 2020, 136, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, K.; Oikawa, D.; Iio, K.; Obika, S.; Hori, Y.; Urashima, T.; Ayukawa, K.; Tokunaga, F. Small-Molecule Inhibitors of Linear Ubiquitin Chain Assembly Complex (LUBAC), HOIPINs, Suppress NF-ΚB Signaling. Biochem. Biophys. Res. Commun. 2019, 509, 700–706. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, V.; Johnson, C.; Barlow, V.; Hastie, J.; Knebel, A.; Trost, M. The MALDI-TOF E2/E3 Ligase Assay as Universal Tool for Drug Discovery in the Ubiquitin Pathway. Cell Chem. Biol. 2018, 25, 1117–1127.e4. [Google Scholar] [CrossRef] [PubMed]
- Bogeljić Patekar, M.; Milunović, V.; Mišura Jakobac, K.; Perica, D.; Mandac Rogulj, I.; Kursar, M.; Planinc-Peraica, A.; Ostojić Kolonić, S. Bendamustine: An old drug in the new era for patients with non-hodgkin lymphomas and chronic lymphocytic leukemia. Acta Clin. Croat. 2018, 57, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Damaj, G.; Gressin, R.; Bouabdallah, K.; Cartron, G.; Choufi, B.; Gyan, E.; Banos, A.; Jaccard, A.; Park, S.; Tournilhac, O.; et al. Results from a Prospective, Open-Label, Phase II Trial of Bendamustine in Refractory or Relapsed T-Cell Lymphomas: The BENTLY Trial. J. Clin. Oncol. 2013, 31, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, E.; Neumann, A.; Fauth, F.; Atmaca, A.; Al-Batran, S.E.; Pauligk, C.; Jäger, E. Phase II Study of Bendamustine in Combination with Rituximab as First-Line Treatment in Patients 80 Years or Older with Aggressive B-Cell Lymphomas. Ann. Oncol. 2011, 22, 1839–1844. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the Chains: Structure and Function of the Deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Di Lello, P.; Pastor, R.; Murray, J.M.; Blake, R.A.; Cohen, F.; Crawford, T.D.; Drobnick, J.; Drummond, J.; Kategaya, L.; Kleinheinz, T.; et al. Discovery of Small-Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and in Silico Techniques. J. Med. Chem. 2017, 60, 10056–10070. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Paner, A.; Berdeja, J.G.; Paba-Prada, C.; Venugopal, P.; Porkka, K.; Gullbo, J.; Linder, S.; Loskog, A.; Richardson, P.G.; et al. Phase 1 Study of the Protein Deubiquitinase Inhibitor VLX1570 in Patients with Relapsed and/or Refractory Multiple Myeloma. Investig. New Drugs 2020, 38, 1448–1453. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Tuckett, A.Z.; Ouk, S.; Liou, H.-C.; Altan-Bonnet, G.; Tsai, J.J.; Oyler, J.E.; Smith, O.M.; West, M.L.; Singer, N.V.; et al. A Small-Molecule c-Rel Inhibitor Reduces Alloactivation of T Cells without Compromising Antitumor Activity. Cancer Discov. 2014, 4, 578–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shono, Y.; Tuckett, A.Z.; Liou, H.-C.; Doubrovina, E.; Derenzini, E.; Ouk, S.; Tsai, J.J.; Smith, O.M.; Levy, E.R.; Kreines, F.M.; et al. Characterization of a C-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-ΚB-Controlled Oxidative Stress Responses. Cancer Res. 2016, 76, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Vaisitti, T.; Gaudino, F.; Ouk, S.; Moscvin, M.; Vitale, N.; Serra, S.; Arruga, F.; Zakrzewski, J.L.; Liou, H.-C.; Allan, J.N.; et al. Targeting Metabolism and Survival in Chronic Lymphocytic Leukemia and Richter Syndrome Cells by a Novel NF-ΚB Inhibitor. Haematologica 2017, 102, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Shrum, C.K.; Defrancisco, D.; Meffert, M.K. Stimulated Nuclear Translocation of NF-KappaB and Shuttling Differentially Depend on Dynein and the Dynactin Complex. Proc. Natl. Acad. Sci. USA 2009, 106, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- Valovka, T.; Hottiger, M.O. P65 Controls NF-ΚB Activity by Regulating Cellular Localization of IκBβ. Biochem. J. 2011, 434, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, R.; Kinnunen, L.; Köhler, M.; Julkunen, I.; Melén, K. NF-{kappa}B Is Transported into the Nucleus by Importin {alpha}3 and Importin {alpha}4. J. Biol. Chem. 2005, 280, 15942–15951. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, B.; Li, T.; Peng, Y.; Guan, J.; Lai, S.; Zheng, X. A CRM1-Dependent Nuclear Export Signal Controls Nucleocytoplasmic Translocation of HSCARG, Which Regulates NF-ΚB Activity. Traffic 2012, 13, 790–799. [Google Scholar] [CrossRef]
- Ferreira, B.I.; Cautain, B.; Grenho, I.; Link, W. Small Molecule Inhibitors of CRM1. Front. Pharmacol. 2020, 11, 625. [Google Scholar] [CrossRef]
- Rahmani, K.; Dean, D.A. Leptomycin B Alters the Subcellular Distribution of CRM1 (Exportin 1). Biochem. Biophys. Res. Commun. 2017, 488, 253–258. [Google Scholar] [CrossRef]
- Sakakibara, K.; Saito, N.; Sato, T.; Suzuki, A.; Hasegawa, Y.; Friedman, J.M.; Kufe, D.W.; Vonhoff, D.D.; Iwami, T.; Kawabe, T. CBS9106 Is a Novel Reversible Oral CRM1 Inhibitor with CRM1 Degrading Activity. Blood 2011, 118, 3922–3931. [Google Scholar] [CrossRef] [PubMed]
- Cornell, R.F.; Baz, R.; Richter, J.R.; Rossi, A.; Vogl, D.T.; Chen, C.; Shustik, C.; Alvarez, M.J.; Shen, Y.; Unger, T.J.; et al. A Phase 1 Clinical Trial of Oral Eltanexor in Patients with Relapsed or Refractory Multiple Myeloma. Am. J. Hematol. 2022, 97, E54–E58. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Z.; Yao, S.Y.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of Nuclear Translocation of Transcription Factor NF-Kappa B by a Synthetic Peptide Containing a Cell Membrane-Permeable Motif and Nuclear Localization Sequence. J. Biol. Chem. 1995, 270, 14255–14258. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Liu, B.; Cheng, Y.; Qian, H.; Yang, H.; Li, X.; Yang, G.; Zheng, X.; Shen, F. SN50 Attenuates Alveolar Hypercoagulation and Fibrinolysis Inhibition in Acute Respiratory Distress Syndrome Mice through Inhibiting NF-ΚB P65 Translocation. Respir. Res. 2020, 21, 130. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin Is a Specific Inhibitor of Importin α/β-Mediated Nuclear Import Able to Inhibit Replication of HIV-1 and Dengue Virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a Small Molecule Inhibitor of the Transport Receptor Importin-β. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Xu, X.; Fan, X.; Zhang, C.; Wei, Q.; Wang, X.; Guo, W.; Xing, W.; Yu, J.; Yan, J.-L.; et al. A Cell-Penetrating Peptide Suppresses Inflammation by Inhibiting NF-ΚB Signaling. Mol. Ther. 2011, 19, 1849–1857. [Google Scholar] [CrossRef]
- Horie, K.; Ma, J.; Umezawa, K. Inhibition of Canonical NF-ΚB Nuclear Localization by (-)-DHMEQ via Impairment of DNA Binding. Oncol. Res. 2015, 22, 105–115. [Google Scholar] [CrossRef]
- Horie, R.; Watanabe, M.; Okamura, T.; Taira, M.; Shoda, M.; Motoji, T.; Utsunomiya, A.; Watanabe, T.; Higashihara, M.; Umezawa, K. DHMEQ, a New NF-KappaB Inhibitor, Induces Apoptosis and Enhances Fludarabine Effects on Chronic Lymphocytic Leukemia Cells. Leukemia 2006, 20, 800–806. [Google Scholar] [CrossRef]
- Matsumoto, G.; Namekawa, J.; Muta, M.; Nakamura, T.; Bando, H.; Tohyama, K.; Toi, M.; Umezawa, K. Targeting of Nuclear Factor KappaB Pathways by Dehydroxymethylepoxyquinomicin, a Novel Inhibitor of Breast Carcinomas: Antitumor and Antiangiogenic Potential in Vivo. Clin. Cancer Res. 2005, 11, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Lach, M.S.; Borska, S.; Nowakowski, B.; Umezawa, K.; Suchorska, W.M. DHMEQ Enhances the Cytotoxic Effect of Cisplatin and Carboplatin in Ovarian Cancer Cell Lines. Am. J. Cancer Res. 2021, 11, 6024–6041. [Google Scholar] [PubMed]
- Ryan, S.-L.; Beard, S.; Barr, M.P.; Umezawa, K.; Heavey, S.; Godwin, P.; Gray, S.G.; Cormican, D.; Finn, S.P.; Gately, K.A.; et al. Targeting NF-ΚB-Mediated Inflammatory Pathways in Cisplatin-Resistant NSCLC. Lung Cancer 2019, 135, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.-D.; Lamb, A.; Chen, L.-F. Posttranslational Modifications of NF-KappaB: Another Layer of Regulation for NF-KappaB Signaling Pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Collares, D. Post-Translational Modifications of RelB NF-ΚB Subunit and Associated Functions. Cells 2016, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-ΚB P65 and Strategies for Therapeutic Manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef]
- Kim, N.; Norsworthy, K.J.; Subramaniam, S.; Chen, H.; Manning, M.L.; Kitabi, E.; Earp, J.; Ehrlich, L.A.; Okusanya, O.O.; Vallejo, J.; et al. FDA Approval Summary: Decitabine and Cedazuridine Tablets for Myelodysplastic Syndromes. Clin. Cancer Res. 2022, 28, 3411–3416. [Google Scholar] [CrossRef]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Azacitidine (5-Azacytidine, Vidaza) for Injectable Suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef]
- Wu, D.; Qiu, Y.; Jiao, Y.; Qiu, Z.; Liu, D. Small Molecules Targeting HATs, HDACs, and BRDs in Cancer Therapy. Front. Oncol. 2020, 10, 560487. [Google Scholar] [CrossRef]
- Kim, H.-J.; Bae, S.-C. Histone Deacetylase Inhibitors: Molecular Mechanisms of Action and Clinical Trials as Anti-Cancer Drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, Depsipeptide): A Natural Product Recently Approved for Cutaneous T-Cell Lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Su, Y.; Hege, K.; Madlambayan, G.; Edwards, H.; Knight, T.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; et al. The HDAC and PI3K Dual Inhibitor CUDC-907 Synergistically Enhances the Antileukemic Activity of Venetoclax in Preclinical Models of Acute Myeloid Leukemia. Haematologica 2021, 106, 1262–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Bian, X.; Lin, W. The Dual HDAC-PI3K Inhibitor CUDC-907 Displays Single-Agent Activity and Synergizes with PARP Inhibitor Olaparib in Small Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 219. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Kelly, K.R.; Flinn, I.; Patel, M.R.; Gharavi, R.; Ma, A.; Parker, J.; Hafeez, A.; Tuck, D.; Younes, A. CUDC-907 in Relapsed/Refractory Diffuse Large B-Cell Lymphoma, Including Patients with MYC-Alterations: Results from an Expanded Phase I Trial. Haematologica 2017, 102, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFκB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.-F. Brd4 Maintains Constitutively Active NF-ΚB in Cancer Cells by Binding to Acetylated RelA. Oncogene 2014, 33, 2395–2404. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain Inhibitor OTX015 in Patients with Acute Leukaemia: A Dose-Escalation, Phase 1 Study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain Inhibitor OTX015 in Patients with Lymphoma or Multiple Myeloma: A Dose-Escalation, Open-Label, Pharmacokinetic, Phase 1 Study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Braña, I.; Cassier, P.A.; Moreno, V.; de Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr. 2020, 4, pkz093. [Google Scholar] [CrossRef]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET Protein Proteolysis-Targeting Chimera Exerts Superior Lethal Activity than Bromodomain Inhibitor (BETi) against Post-Myeloproliferative Neoplasm Secondary (s) AML Cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Fiskus, W.; Qian, Y.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET Protein Proteolysis Targeting Chimera (PROTAC) Exerts Potent Lethal Activity against Mantle Cell Lymphoma Cells. Leukemia 2018, 32, 343–352. [Google Scholar] [CrossRef] [PubMed]
- García-Piñeres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in P65/NF-KappaB Plays a Crucial Role in DNA Binding Inhibition by Sesquiterpene Lactones. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [PubMed]
- Sztiller-Sikorska, M.; Czyz, M. Parthenolide as Cooperating Agent for Anti-Cancer Treatment of Various Malignancies. Pharmaceuticals 2020, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Paço, A.; Brás, T.; Santos, J.O.; Sampaio, P.; Gomes, A.C.; Duarte, M.F. Anti-Inflammatory and Immunoregulatory Action of Sesquiterpene Lactones. Molecules 2022, 27, 1142. [Google Scholar] [CrossRef] [PubMed]
- Mendonca, M.S.; Turchan, W.T.; Alpuche, M.E.; Watson, C.N.; Estabrook, N.C.; Chin-Sinex, H.; Shapiro, J.B.; Imasuen-Williams, I.E.; Rangel, G.; Gilley, D.P.; et al. DMAPT Inhibits NF-ΚB Activity and Increases Sensitivity of Prostate Cancer Cells to X-Rays in Vitro and in Tumor Xenografts in Vivo. Free Radic. Biol. Med. 2017, 112, 318–326. [Google Scholar] [CrossRef]
- Lamture, G.; Crooks, P.A.; Borrelli, M.J. Actinomycin-D and Dimethylamino-Parthenolide Synergism in Treating Human Pancreatic Cancer Cells. Drug Dev. Res. 2018, 79, 287–294. [Google Scholar] [CrossRef]
- Deraska, P.V.; O’Leary, C.; Reavis, H.D.; Labe, S.; Dinh, T.-K.; Lazaro, J.-B.; Sweeney, C.; D’Andrea, A.D.; Kozono, D. NF-ΚB Inhibition by Dimethylaminoparthenolide Radiosensitizes Non-Small-Cell Lung Carcinoma by Blocking DNA Double-Strand Break Repair. Cell Death Discov. 2018, 4, 10. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Porciani, D.; Tedeschi, L.; Marchetti, L.; Citti, L.; Piazza, V.; Beltram, F.; Signore, G. Aptamer-Mediated Codelivery of Doxorubicin and NF-ΚB Decoy Enhances Chemosensitivity of Pancreatic Tumor Cells. Mol. Ther. Nucleic Acids 2015, 4, e235. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Sandomenico, A.; Raimondo, D.; Low, C.; Rocci, A.; Tralau-Stewart, C.; Capece, D.; D’Andrea, D.; Bua, M.; Boyle, E.; et al. Cancer-Selective Targeting of the NF-ΚB Survival Pathway with GADD45β/MKK7 Inhibitors. Cancer Cell 2014, 26, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; De Smaele, E.; Zazzeroni, F.; Nguyen, D.U.; Papa, S.; Jones, J.; Cox, C.; Gelinas, C.; Franzoso, G. Regulation of the Gadd45beta Promoter by NF-KappaB. DNA Cell Biol. 2002, 21, 491–503. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Zazzeroni, F.; Papa, S.; Nguyen, D.U.; Jin, R.; Jones, J.; Cong, R.; Franzoso, G. Induction of Gadd45beta by NF-KappaB Downregulates pro-Apoptotic JNK Signalling. Nature 2001, 414, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J.J. Gadd45 in Stress Signaling, Cell Cycle Control, and Apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Liu, Y. GADD45 Proteins: Roles in Cellular Senescence and Tumor Development. Exp. Biol. Med. (Maywood) 2014, 239, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, L.; Capece, D.; D’Andrea, D.; Begalli, F.; Verzella, D.; Bennett, J.; Acton, G.; Campbell, E.A.; Kelly, J.; Tarbit, M.; et al. Clinical Proof of Concept for a Safe and Effective NF-ΚB-Targeting Strategy in Multiple Myeloma. Br. J. Haematol. 2019, 185, 588–592. [Google Scholar] [CrossRef]
- Tornatore, L.; Capece, D.; D’Andrea, D.; Begalli, F.; Verzella, D.; Bennett, J.; Acton, G.; Campbell, E.A.; Kelly, J.; Tarbit, M.; et al. Preclinical Toxicology and Safety Pharmacology of the First-in-Class GADD45β/MKK7 Inhibitor and Clinical Candidate, DTP3. Toxicol. Rep. 2019, 6, 369–379. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.F.; Zamyatnin, A.A.; Soond, S.M. BH3-Mimetics: Recent Developments in Cancer Therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355. [Google Scholar] [CrossRef]
- Suvarna, V.; Singh, V.; Murahari, M. Current Overview on the Clinical Update of Bcl-2 Anti-Apoptotic Inhibitors for Cancer Therapy. Eur. J. Pharmacol. 2019, 862, 172655. [Google Scholar] [CrossRef]
- Juárez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to Date and Clinical Potential. Drugs Context 2019, 8, 212574. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, B.; Verzella, D.; Capece, D.; Vecchiotti, D.; Di Vito Nolfi, M.; Flati, I.; Cornice, J.; Di Padova, M.; Angelucci, A.; Alesse, E.; et al. NF-ΚB: A Druggable Target in Acute Myeloid Leukemia. Cancers 2022, 14, 3357. [Google Scholar] [CrossRef] [PubMed]
- Sidiqi, M.H.; Al Saleh, A.S.; Kumar, S.K.; Leung, N.; Jevremovic, D.; Muchtar, E.; Gonsalves, W.I.; Kourelis, T.V.; Warsame, R.; Buadi, F.K.; et al. Venetoclax for the Treatment of Multiple Myeloma: Outcomes Outside of Clinical Trials. Am. J. Hematol. 2021, 96, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.; Letai, A. Why Do BCL-2 Inhibitors Work and Where Should We Use Them in the Clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-Quadruplexes: A Promising Target for Cancer Therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Liu, Z.; Wild, C.; Ding, Y.; Ye, N.; Chen, H.; Wold, E.A.; Zhou, J. BH4 Domain of Bcl-2 as a Novel Target for Cancer Therapy. Drug Discov. Today 2016, 21, 989–996. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and Its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-Angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without Bevacizumab in Unresectable Hepatocellular Carcinoma (GO30140): An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.; Merle, P.; et al. Updated Efficacy and Safety Data from IMbrave150: Atezolizumab plus Bevacizumab vs Sorafenib for Unresectable Hepatocellular Carcinoma. J. Hepatol. 2022, 76, 862–873. [Google Scholar] [CrossRef]
- Blumenthal, G.M.; Cortazar, P.; Zhang, J.J.; Tang, S.; Sridhara, R.; Murgo, A.; Justice, R.; Pazdur, R. FDA Approval Summary: Sunitinib for the Treatment of Progressive Well-Differentiated Locally Advanced or Metastatic Pancreatic Neuroendocrine Tumors. Oncologist 2012, 17, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Yoshioka, H.; Oshita, F.; Hida, T.; Yoh, K.; Hayashi, H.; Kato, T.; Kaneda, H.; Yamada, K.; Tanaka, H.; et al. Phase III, Randomized, Placebo-Controlled, Double-Blind Trial of Motesanib (AMG-706) in Combination With Paclitaxel and Carboplatin in East Asian Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2017, 35, 3662–3670. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Morden, J.P.; Kilburn, L.; Leahy, M.; Benson, C.; Bhadri, V.; Campbell-Hewson, Q.; Cubedo, R.; Dangoor, A.; Fox, L.; et al. Cediranib in Patients with Alveolar Soft-Part Sarcoma (CASPS): A Double-Blind, Placebo-Controlled, Randomised, Phase 2 Trial. Lancet Oncol. 2019, 20, 1023–1034. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Johnson, D.H.; Abdel-Wahab, N.; Foo, W.C.; Bentebibel, S.-E.; Daher, M.; Haymaker, C.; Wani, K.; Saberian, C.; Ogata, D.; et al. Interleukin-6 Blockade Abrogates Immunotherapy Toxicity and Promotes Tumor Immunity. Cancer Cell 2022, 40, 509–523.e6. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Abdel-Wahab, N.; Bentebibel, S.E.; Montazari, E.J.; Spillson, C.A.; Ekmekcioglu, S.; Gao, J.; Altan, M. 169TiP Tocilizumab, Ipilimumab, and Nivolumab for the Treatment of Advanced Melanoma, Non-Small Cell Lung Cancer, or Urothelial Carcinoma. Ann. Oncol. 2021, 32, S1456. [Google Scholar] [CrossRef]
- Capece, D.; Verzella, D.; Di Francesco, B.; Alesse, E.; Franzoso, G.; Zazzeroni, F. NF-ΚB and Mitochondria Cross Paths in Cancer: Mitochondrial Metabolism and Beyond. Semin. Cell Dev. Biol. 2020, 98, 118–128. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic Targeting of “Undruggable” MYC. EBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef]
- Reid, M.A.; Lowman, X.H.; Pan, M.; Tran, T.Q.; Warmoes, M.O.; Ishak Gabra, M.B.; Yang, Y.; Locasale, J.W.; Kong, M. IKKβ Promotes Metabolic Adaptation to Glutamine Deprivation via Phosphorylation and Inhibition of PFKFB3. Genes Dev. 2016, 30, 1837–1851. [Google Scholar] [CrossRef]
- Capece, D.; D’Andrea, D.; Begalli, F.; Goracci, L.; Tornatore, L.; Alexander, J.L.; Di Veroli, A.; Leow, S.-C.; Vaiyapuri, T.S.; Ellis, J.K.; et al. Enhanced Triacylglycerol Catabolism by Carboxylesterase 1 Promotes Aggressive Colorectal Carcinoma. J. Clin. Investig. 2021, 131, e137845. [Google Scholar] [CrossRef]
- Capece, D.; Franzoso, G. Rewired Lipid Metabolism as an Actionable Vulnerability of Aggressive Colorectal Carcinoma. Mol. Cell. Oncol. 2022, 9, 2024051. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-ΚB: Blending Metabolism, Immunity, and Inflammation. Trends Immunol. 2022, 43, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.; Ben Ali, Y.; Lyon, J.; Wang, H.; Nelson, R.; Dolinsky, V.W.; Dyck, J.R.B.; Mitchell, G.; Korbutt, G.S.; Lehner, R. Loss of TGH/Ces3 in Mice Decreases Blood Lipids, Improves Glucose Tolerance, and Increases Energy Expenditure. Cell Metab. 2010, 11, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verzella, D.; Cornice, J.; Arboretto, P.; Vecchiotti, D.; Di Vito Nolfi, M.; Capece, D.; Zazzeroni, F.; Franzoso, G. The NF-κB Pharmacopeia: Novel Strategies to Subdue an Intractable Target. Biomedicines 2022, 10, 2233. https://doi.org/10.3390/biomedicines10092233
Verzella D, Cornice J, Arboretto P, Vecchiotti D, Di Vito Nolfi M, Capece D, Zazzeroni F, Franzoso G. The NF-κB Pharmacopeia: Novel Strategies to Subdue an Intractable Target. Biomedicines. 2022; 10(9):2233. https://doi.org/10.3390/biomedicines10092233
Chicago/Turabian StyleVerzella, Daniela, Jessica Cornice, Paola Arboretto, Davide Vecchiotti, Mauro Di Vito Nolfi, Daria Capece, Francesca Zazzeroni, and Guido Franzoso. 2022. "The NF-κB Pharmacopeia: Novel Strategies to Subdue an Intractable Target" Biomedicines 10, no. 9: 2233. https://doi.org/10.3390/biomedicines10092233
APA StyleVerzella, D., Cornice, J., Arboretto, P., Vecchiotti, D., Di Vito Nolfi, M., Capece, D., Zazzeroni, F., & Franzoso, G. (2022). The NF-κB Pharmacopeia: Novel Strategies to Subdue an Intractable Target. Biomedicines, 10(9), 2233. https://doi.org/10.3390/biomedicines10092233