
Androgen-Independent Prostate Cancer Is Sensitive to CDC42-PAK7 Kinase Inhibition

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Isolation and MicroRNA Microarray
2.3. RNA-Sequencing Data Analysis
2.4. RT-PCR
2.5. In Silico Drug Sensitivity and Genetic Perturbation Sensitivity Screening
2.6. Target Validation in Cell Lines Using Western Blots
2.7. QKI-Overexpression by CRISPR/dCas9 System
2.8. In Vivo Tumor Xenograft and Drug Treatment
2.9. Collection of Human Prostate Cancer Samples and Immunohistochemistry
2.10. RNA Interferences and Drug Treatment In Vitro
2.11. Statistical Analyses
3. Results
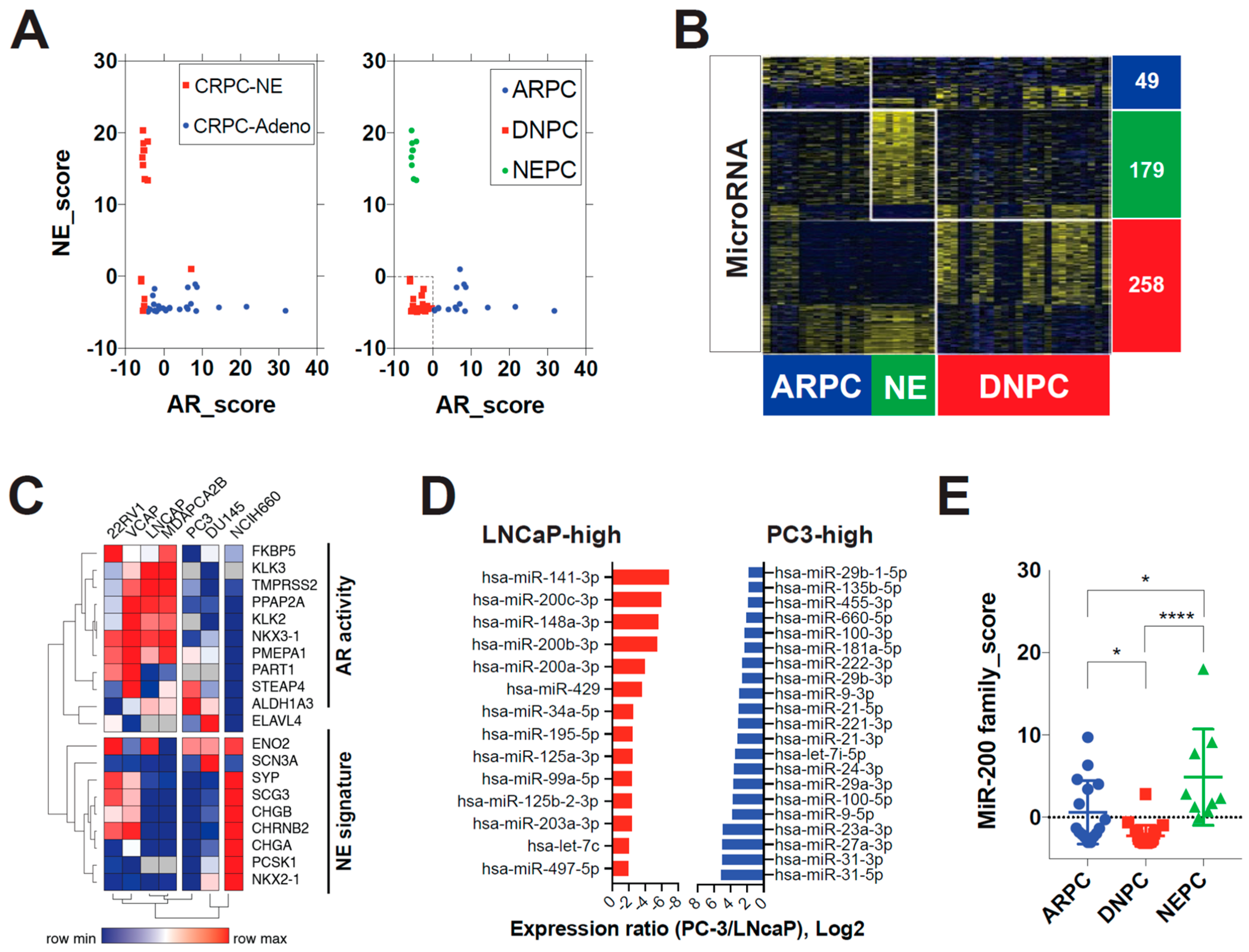
3.1. MicroRNA-200 Family Are Downregulated in Double-Negative Prostate Cancer (DNPC)
3.2. QKI Is a MicroRNA-200 Family Target Gene That Is Overexpressed in Post-ADT Prostate Tumors
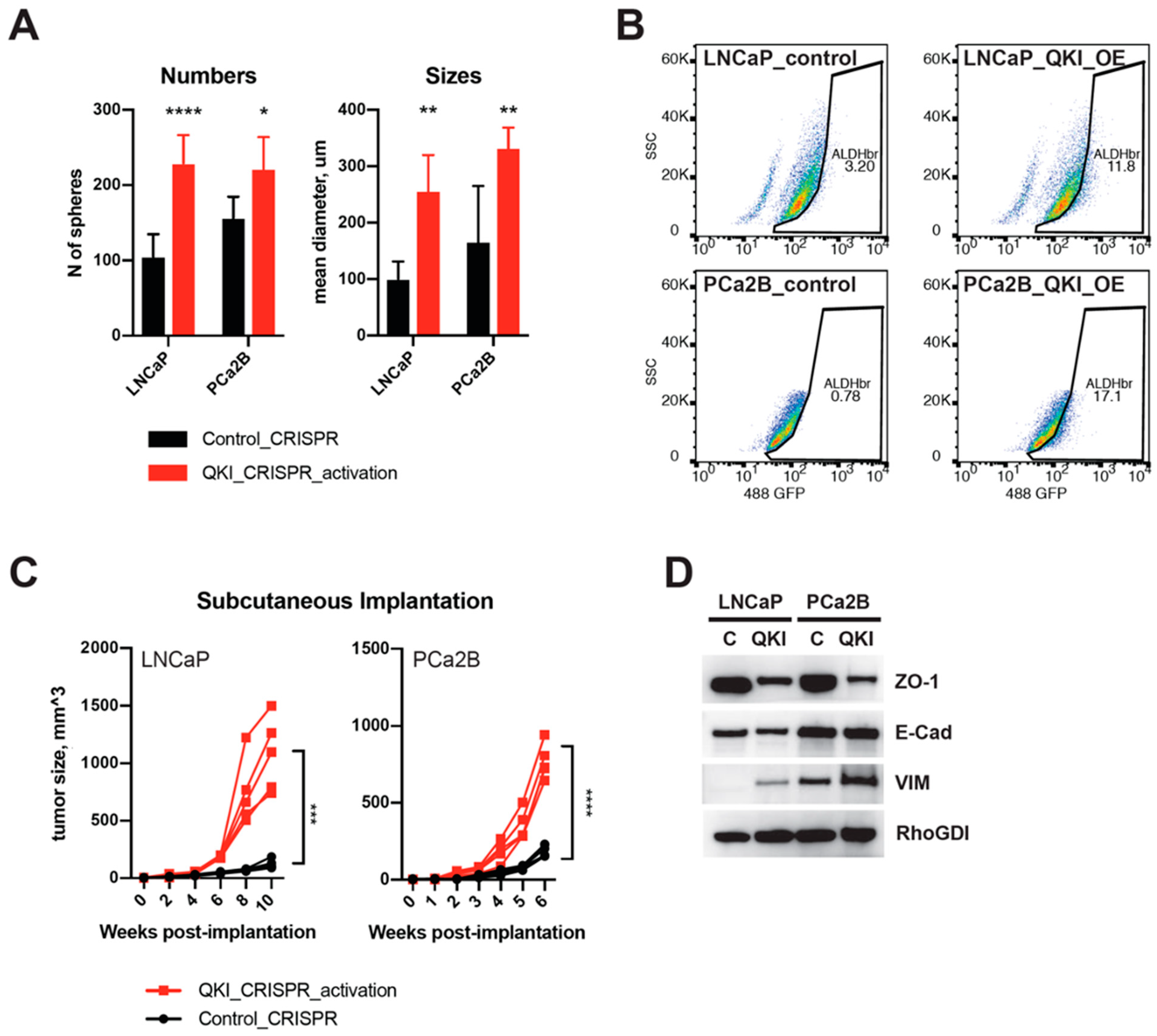
3.3. QKI Overexpression in Prostate Cancer Drives Tumor Progression and Epithelial-to-Mesenchymal Transition
3.4. QKI Knockdown Decreases Expressions of Cell Cycle and EMT Genes in Prostate Cancer Cell Line
3.5. Characterization of a Therapy-Resistant Mesenchymal-like State and the Impact of GPX4 Inhibitors and Statins in Tumor Cells
3.6. Fluvastatin Inhibits the Growth of AR-Lost Castration-Resistant Prostate Cancer Tumors by Targeting the QKI and CDC42 Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Lowrance, W.T.; Murad, M.H.; Oh, W.K.; Jarrard, D.F.; Resnick, M.J.; Cookson, M.S. Castration-resistant prostate cancer: Aua guideline amendment 2018. J. Urol. 2018, 200, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Small, E.J.; Huang, J.T.; Youngren, J.; Sokolov, A.; Aggarwal, R.R.; Thomas, G.; True, L.D.; Zhang, L.; Foye, A.; Alumkal, J.J.; et al. Characterization of neuroendocrine prostate cancer (nepc) in patients with metastatic castration resistant prostate cancer (mcrpc) resistant to abiraterone (abi) or enzalutamide (enz): Preliminary results from the su2c/pcf/aacr west coast prostate cancer dream team (wcdt). J. Clin. Oncol. 2015, 33, 5003. [Google Scholar]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging variants of castration-resistant prostate cancer. Curr. Oncol. Rep. 2017, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, M.H.; Partin, A.W.; Veltri, R.W.; Epstein, J.I. Neuroendocrine differentiation in prostate cancer: Enhanced prediction of progression after radical prostatectomy. Hum. Pathol. 1996, 27, 683–687. [Google Scholar] [CrossRef]
- Ham, W.S.; Cho, N.H.; Kim, W.T.; Ju, H.J.; Lee, J.S.; Choi, Y.D. Pathological effects of prostate cancer correlate with neuroendocrine differentiation and pten expression after bicalutamide monotherapy. J. Urol. 2009, 182, 1378–1384. [Google Scholar] [CrossRef]
- Ismail, A.H.; Landry, F.; Aprikian, A.G.; Chevalier, S. Androgen ablation promotes neuroendocrine cell differentiation in dog and human prostate. Prostate 2002, 51, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.Q.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.M.; et al. Rb1 and trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivas, P.; Bratslavsky, G.; Jacob, J.M.; Kravtsov, O.; Necchi, A.; Spiess, P.E.; Danziger, N.; Lin, D.I.; Huang, R.S.P.; Parini, V.; et al. Association of rb1 mutational status with overall genomic landscape in neuroendocrine prostate cancer (nepc). J. Clin. Oncol. 2022, 40, 5063. [Google Scholar] [CrossRef]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through fgf signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [Green Version]
- Gangaraju, V.K.; Lin, H. Micrornas: Key regulators of stem cells. Nat. Rev. Mol. Cell. Biol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Sharp, P.A. Roles for micrornas in conferring robustness to biological processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebet, B.L.; Mak, R.H.; Ferrando, A.A.; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Porkka, K.P.; Pfeiffer, M.J.; Waltering, K.K.; Vessella, R.L.; Tammela, T.L.J.; Visakorpi, T. Microrna expression profiling in prostate cancer. Cancer Res. 2007, 67, 6130–6135. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, Y.; Feng, W.; Wang, X.; Yang, J.Y.; Zhao, Y.; Wang, Y.; Liu, Y. Transcription factor and microrna regulation in androgen-dependent and -independent prostate cancer cells. BMC Genom. 2008, 9 (Suppl. 2), S22. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kelnar, K.; Vlassov, A.V.; Brown, D.; Wang, J.C.; Tang, D.G. Distinct microrna expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res. 2012, 72, 3393–3404. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microrna target sites in mammalian mrnas. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillman, K.A.; Phillips, C.A.; Roslan, S.; Toubia, J.; Dredge, B.K.; Bert, A.G.; Lumb, R.; Neumann, D.P.; Li, X.; Conn, S.J.; et al. Mir-200/375 control epithelial plasticity-associated alternative splicing by repressing the rna-binding protein quaking. EMBO J. 2018, 37, e99016. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered crispr-cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Humphries, B.; Yang, C.F. The microrna-200 family: Small molecules with novel roles in cancer development, progression and therapy. Oncotarget 2015, 6, 6472–6498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting zeb1 and sip1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Li, T.; Su, Y.; Mei, Y.P.; Leng, Q.X.; Leng, B.J.; Liu, Z.Q.; Stass, S.A.; Jiang, F. Aldh1a1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Burger, P.E.; Gupta, R.; Xiong, X.; Ontiveros, C.S.; Salm, S.N.; Moscatelli, D.; Wilson, E.L. High aldehyde dehydrogenase activity: A novel functional marker of murine prostate stem/progenitor cells. Stem Cells 2009, 27, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.Q.; Park, J.W.; Bebee, T.W.; Warzecha, C.C.; Guo, Y.; Shang, X.Q.; Xing, Y.; Carstens, R.P. Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial-to-mesenchymal transition. Mol. Cell Biol. 2016, 36, 1704–1719. [Google Scholar] [CrossRef] [Green Version]
- Blum, R.; Vethantham, V.; Bowman, C.; Rudnicki, M.; Dynlacht, B.D. Genome-wide identification of enhancers in skeletal muscle: The role of myod1. Genes Dev. 2012, 26, 2763–2779. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Feng, T.; Gao, L.; Sun, F.; Zhou, Q.; Wang, X.; Liu, J.; Zhang, W.; Wang, M.; Xiong, X.; et al. Ppfia4 promotes castration-resistant prostate cancer by enhancing mitochondrial metabolism through mthfd2. J. Exp. Clin. Cancer Res. 2022, 41, 125. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, M.; Liu, Z.; Li, X.; Wang, J.; Fu, N.; Cao, T.; Yang, X. Ppfia4 promotes colon cancer cell proliferation and migration by enhancing tumor glycolysis. Front. Oncol. 2021, 11, 653200. [Google Scholar] [CrossRef] [PubMed]
- Reilich, P.; Krause, S.; Schramm, N.; Klutzny, U.; Bulst, S.; Zehetmayer, B.; Schneiderat, P.; Walter, M.C.; Schoser, B.; Lochmuller, H. A novel mutation in the myotilin gene (myot) causes a severe form of limb girdle muscular dystrophy 1a (lgmd1a). J. Neurol. 2011, 258, 1437–1444. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [Green Version]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid peroxidation-dependent cell death regulated by gpx4 and ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Fish, R.J.; Yang, H.; Viglino, C.; Schorer, R.; Dunoyer-Geindre, S.; Kruithof, E.K.O. Fluvastatin inhibits regulated secretion of endothelial cell von willebrand factor in response to diverse secretagogues. Biochem. J. 2007, 405, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.M.; Elsabrouty, R.; Seemann, J.; Jo, Y.; DeBose-Boyd, R.A. The prenyltransferase ubiad1 is the target of geranylgeraniol in degradation of hmg coa reductase. eLife 2015, 4, e05560. [Google Scholar] [CrossRef] [PubMed]
- Dan, C.; Nath, N.; Liberto, M.; Minden, A. Pak5, a new brain-specific kinase, promotes neurite outgrowth in n1e-115 cells. Mol. Cell Biol. 2002, 22, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Harshman, L.C.; Wang, X.D.; Nakabayashi, M.; Xie, W.L.; Valenca, L.; Werner, L.; Yu, Y.J.; Kantoff, A.M.; Sweeney, C.J.; Mucci, L.A.; et al. Statin use at the time of initiation of androgen deprivation therapy and time to progression in patients with hormone-sensitive prostate cancer. JAMA Oncol. 2015, 1, 495–504. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-fat diet-induced inflammation accelerates prostate cancer growth via il6 signaling. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhang, J.W.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant srebp-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between zeb1 and members of the mir-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Cavallari, I.; Ciccarese, F.; Sharova, E.; Urso, L.; Raimondi, V.; Silic-Benussi, M.; D’Agostino, D.M.; Ciminale, V. The mir-200 family of micrornas: Fine tuners of epithelial-mesenchymal transition and circulating cancer biomarkers. Cancers 2021, 13, 5874. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The mir-200 family determines the epithelial phenotype of cancer cells by targeting the e-cadherin repressors zeb1 and zeb2. Gene Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during emt. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Liao, T.T.; Yang, M.H. Hybrid epithelial/mesenchymal state in cancer metastasis: Clinical significance and regulatory mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldscholte, J.; Berrevoets, C.A.; Risstalpers, C.; Kuiper, G.G.J.M.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in lncap cells contains a mutation in the ligand-binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. 1992, 41, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shi, Y.; Xu, L.L.; Nageswararao, C.; Davis, L.D.; Segawa, T.; Dobi, A.; McLeod, D.G.; Srivastava, S. Androgen receptor mutation (t877a) promotes prostate cancer cell growth and cell survival. Oncogene 2006, 25, 3905–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to map kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium utilization by gpx4 is required to prevent hydroperoxide-induced ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Khatib, A.M.; Siegfried, G.; Prat, A.; Luis, J.; Chretien, M.; Metrakos, P.; Seidah, N.G. Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of ht-29 human colon carcinoma cells—Importance of insulin-like growth factor-1 (igf-1) receptor processing in igf-1-mediated functions. J. Biol. Chem. 2001, 276, 30686–30693. [Google Scholar] [CrossRef] [Green Version]
- Harshman, L.C.; Wang, X.; Nakabayashi, M.; Xie, W.; Valenca, L.; Werner, L.; Yu, Y.; Kantoff, A.M.; Sweeney, C.J.; Mucci, L.A.; et al. Re: Statin use at the time of initiation of androgen deprivation therapy and time to progression in patients with hormone-sensitive prostate cancer editorial comment. J. Urol. 2016, 195, 1780–1781. [Google Scholar]
- Voorneveld, P.W.; Reimers, M.S.; Bastiaannet, E.; Jacobs, R.J.; van Eijk, R.; Zanders, M.M.J.; Herings, R.M.C.; van Herk-Sukel, M.P.P.; Kodach, L.L.; van Wezel, T.; et al. Statin use after diagnosis of colon cancer and patient survival. Gastroenterology 2017, 153, 470–479. [Google Scholar] [CrossRef]
- Pitts, T.M.; Kulikowski, G.N.; Tan, A.C.; Murray, B.W.; Arcaroli, J.J.; Tentler, J.J.; Spreafico, A.; Selby, H.M.; Kachaeva, M.I.; McPhillips, K.L.; et al. Association of the epithelial-to-mesenchymal transition phenotype with responsiveness to the p21-activated kinase inhibitor, pf-3758309, in colon cancer models. Front. Pharmacol. 2013, 4, 35. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.; Park, C.K.; Choi, Y.-D.; Cho, N.H.; Lee, J.; Cho, K.S. Androgen-Independent Prostate Cancer Is Sensitive to CDC42-PAK7 Kinase Inhibition. Biomedicines 2023, 11, 101. https://doi.org/10.3390/biomedicines11010101
Han H, Park CK, Choi Y-D, Cho NH, Lee J, Cho KS. Androgen-Independent Prostate Cancer Is Sensitive to CDC42-PAK7 Kinase Inhibition. Biomedicines. 2023; 11(1):101. https://doi.org/10.3390/biomedicines11010101
Chicago/Turabian StyleHan, Hyunho, Cheol Keun Park, Young-Deuk Choi, Nam Hoon Cho, Jongsoo Lee, and Kang Su Cho. 2023. "Androgen-Independent Prostate Cancer Is Sensitive to CDC42-PAK7 Kinase Inhibition" Biomedicines 11, no. 1: 101. https://doi.org/10.3390/biomedicines11010101
APA StyleHan, H., Park, C. K., Choi, Y.-D., Cho, N. H., Lee, J., & Cho, K. S. (2023). Androgen-Independent Prostate Cancer Is Sensitive to CDC42-PAK7 Kinase Inhibition. Biomedicines, 11(1), 101. https://doi.org/10.3390/biomedicines11010101