

Genetic Ablation of the Nutrient Sensor Ogt in Endocrine Progenitors Is Dispensable for β-Cell Development but Essential for Maintenance of β-Cell Mass

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Blood and Tissue Collection
2.3. Serum Insulin Analysis
2.4. Glucose Tolerance Test
2.5. Tissue Preparation and Immunofluorescence Staining
2.6. Cell Counts and Morphometric Analysis
2.7. Statistical Analysis
3. Results
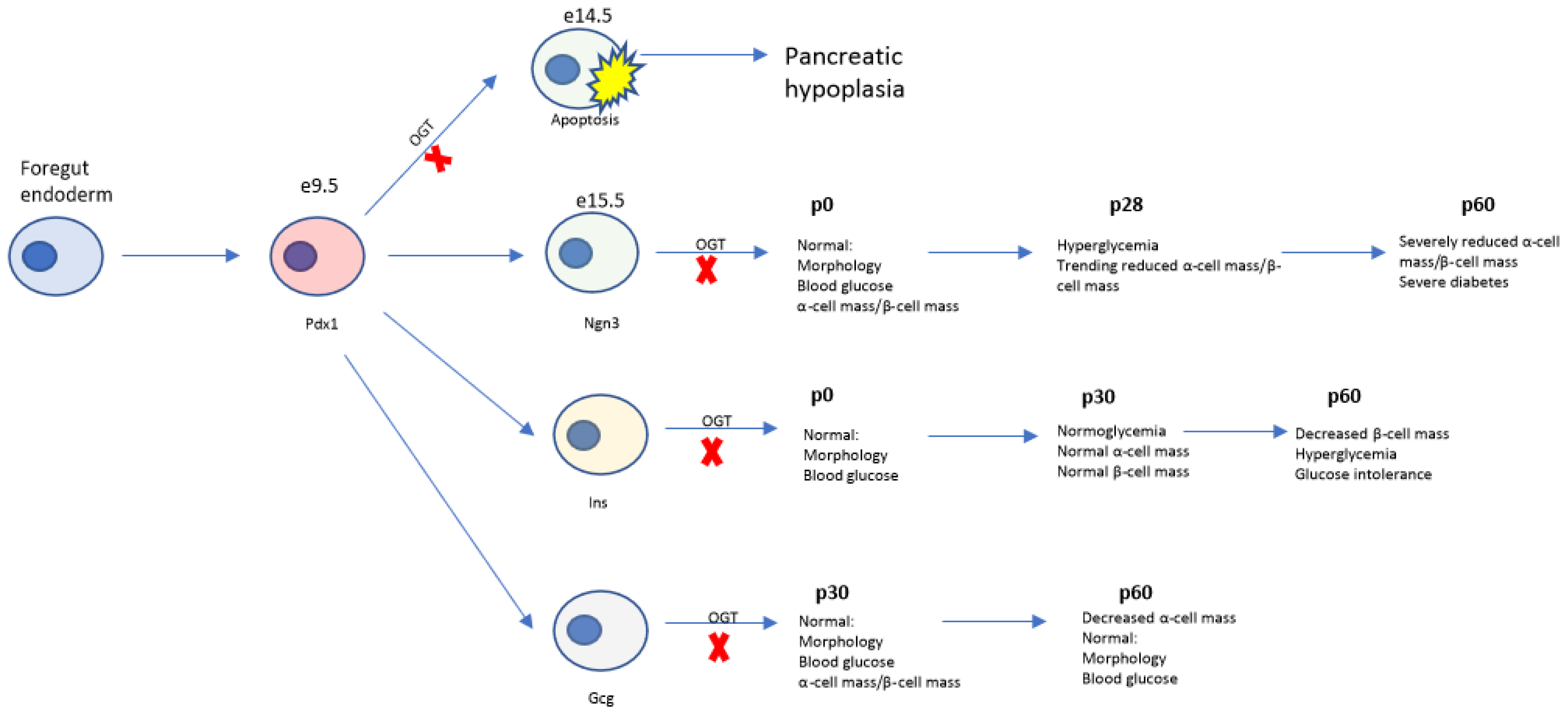
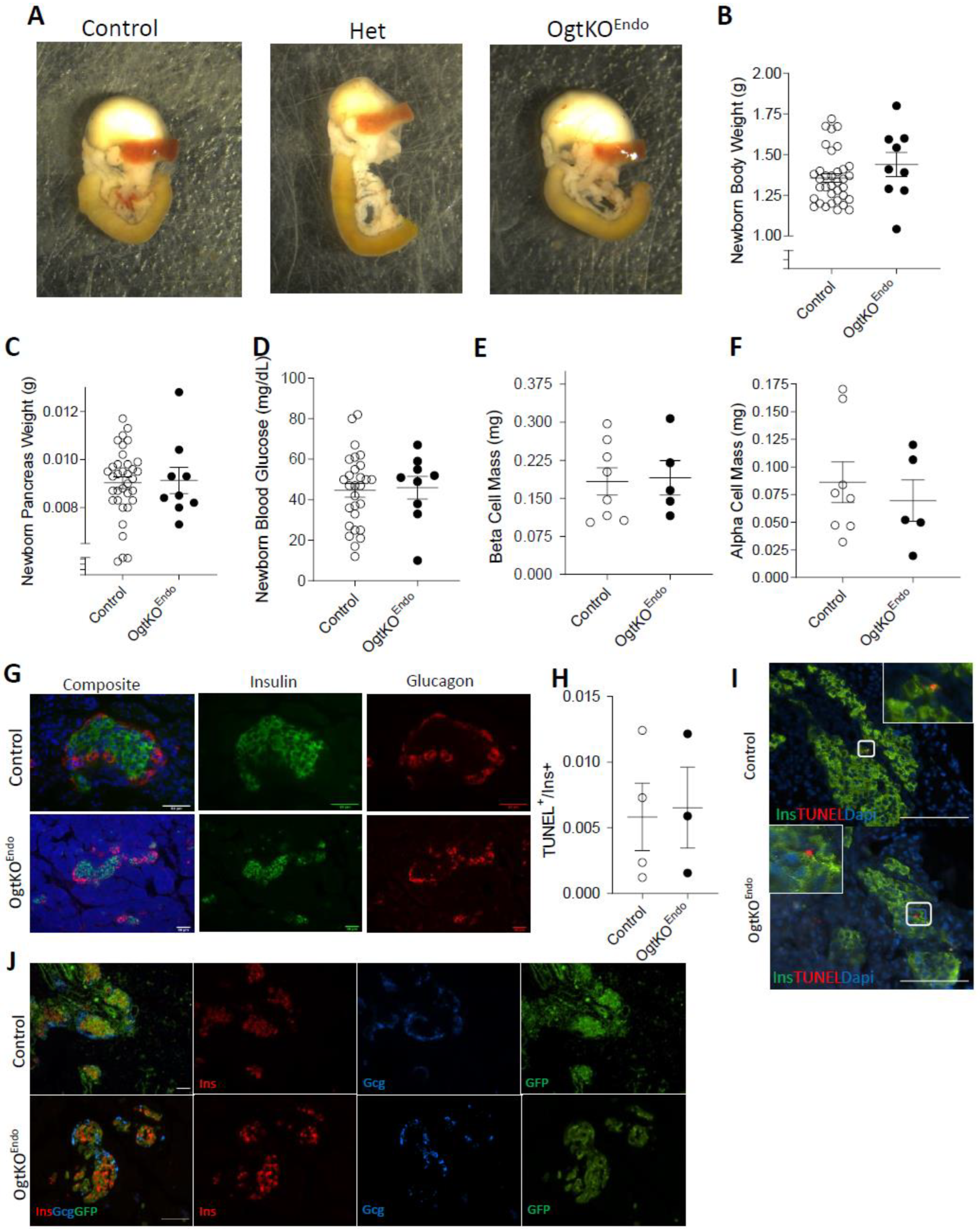
3.1. Deletion of Ogt in the Endocrine Progenitors Has No Effect on Islet Formation
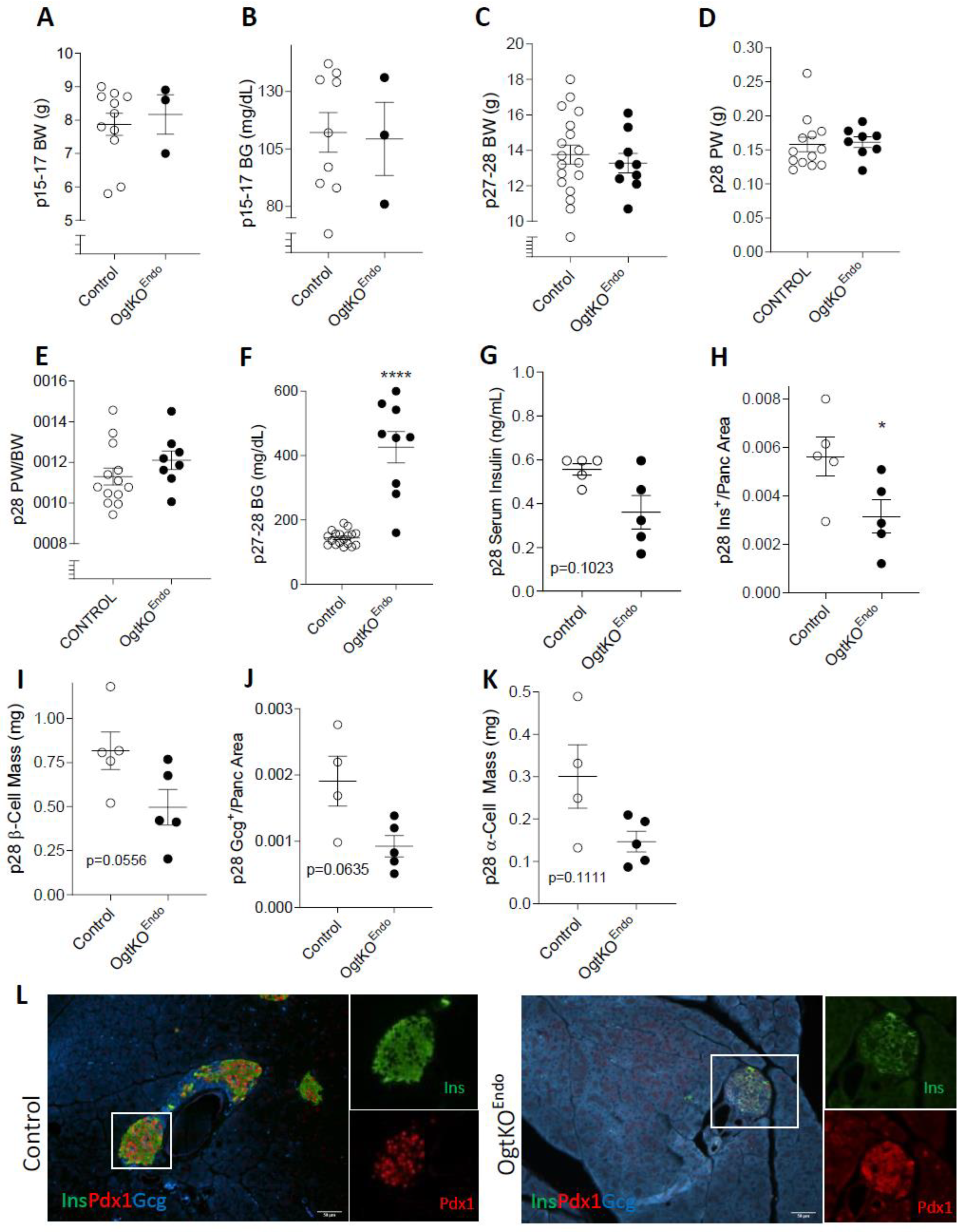
3.2. Progressive Loss of β-Cell Mass and Reduced Immunoreactivity of Pdx1 in OgtKOEndo
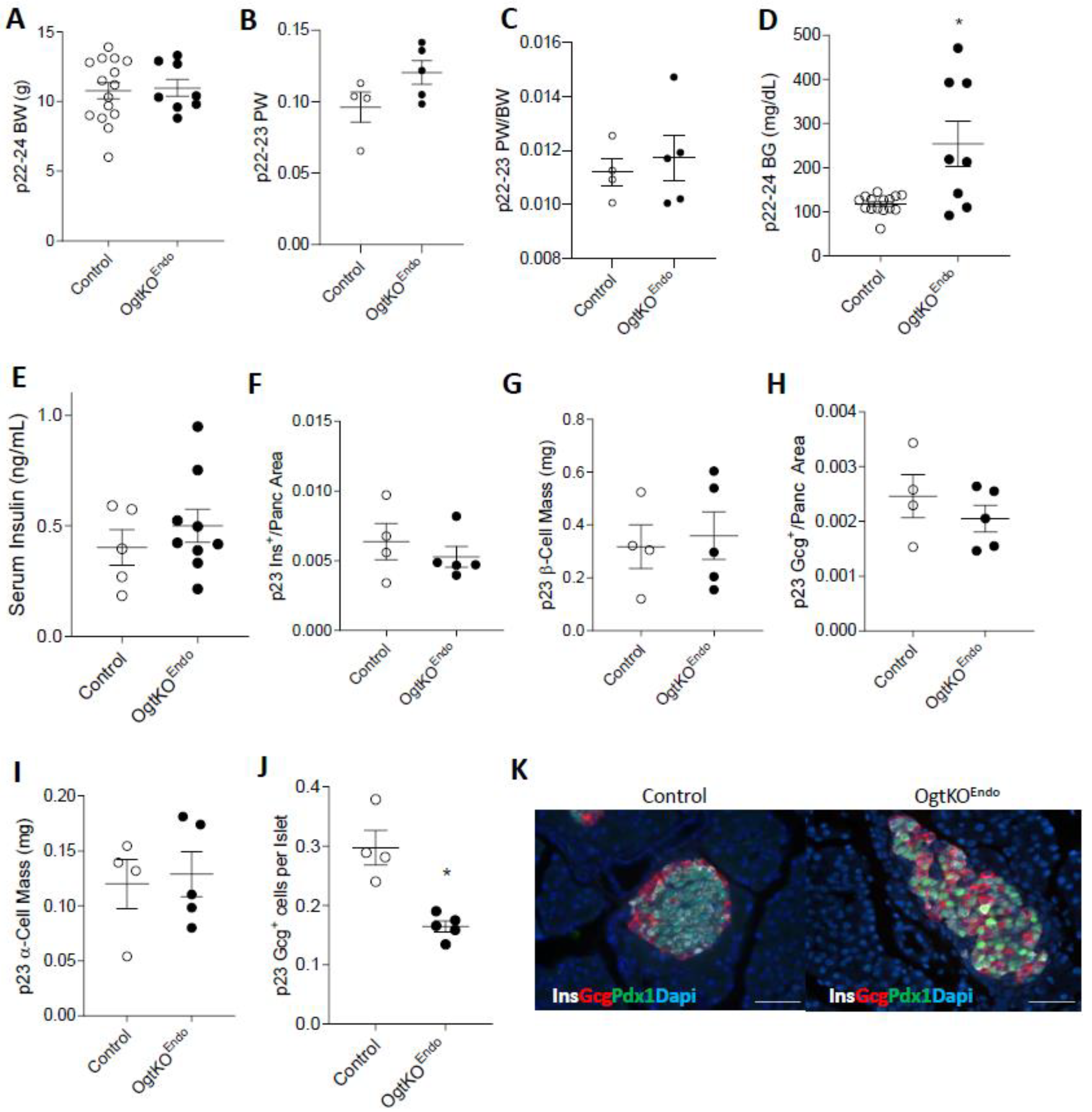
3.3. Aberrations in Islet Architecture in OgtKOEndo at Weaning
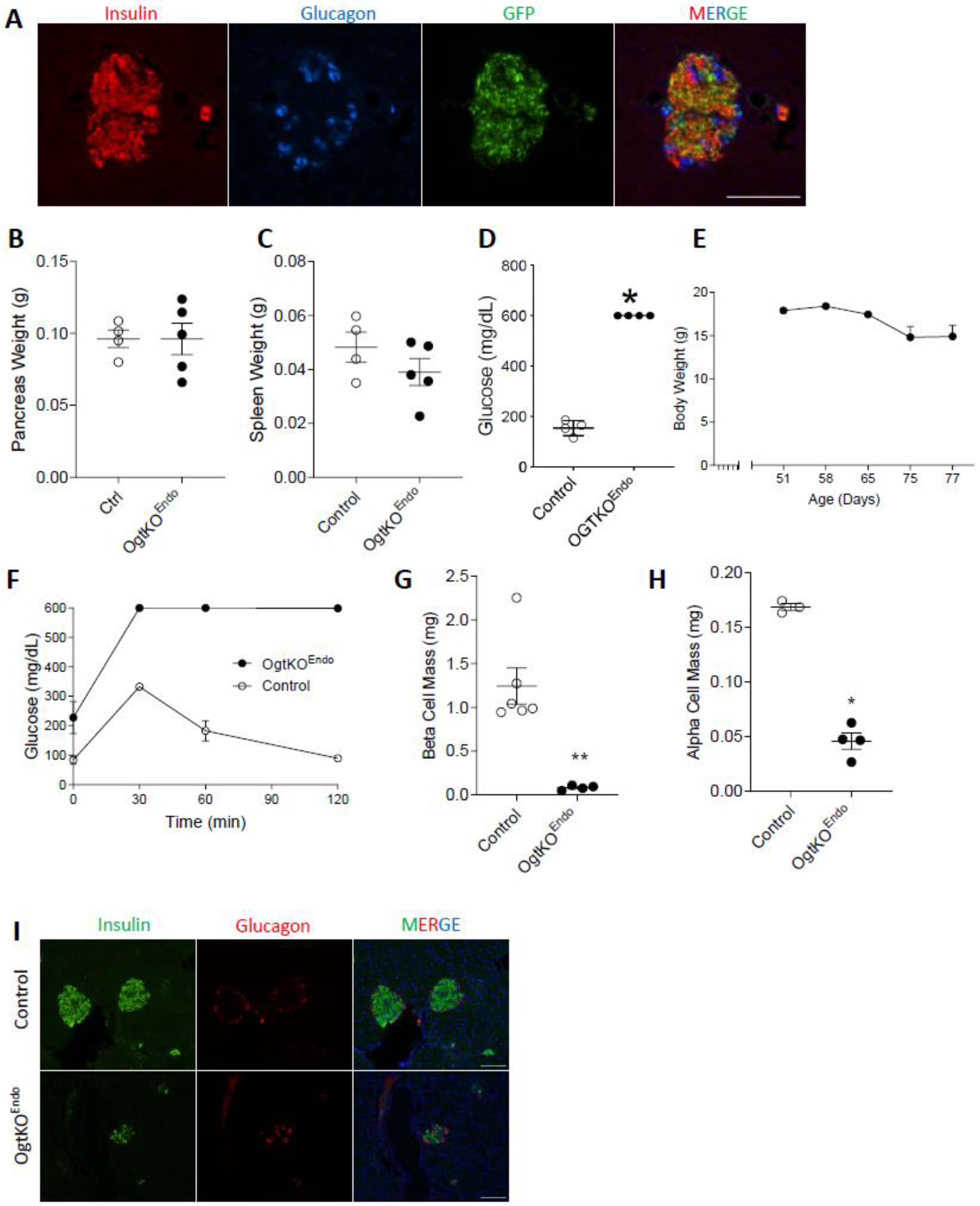
3.4. Adult OgtKOEndo Are Diabetic and Have Both Reduced β-Cell Mass and α-Cell Mass
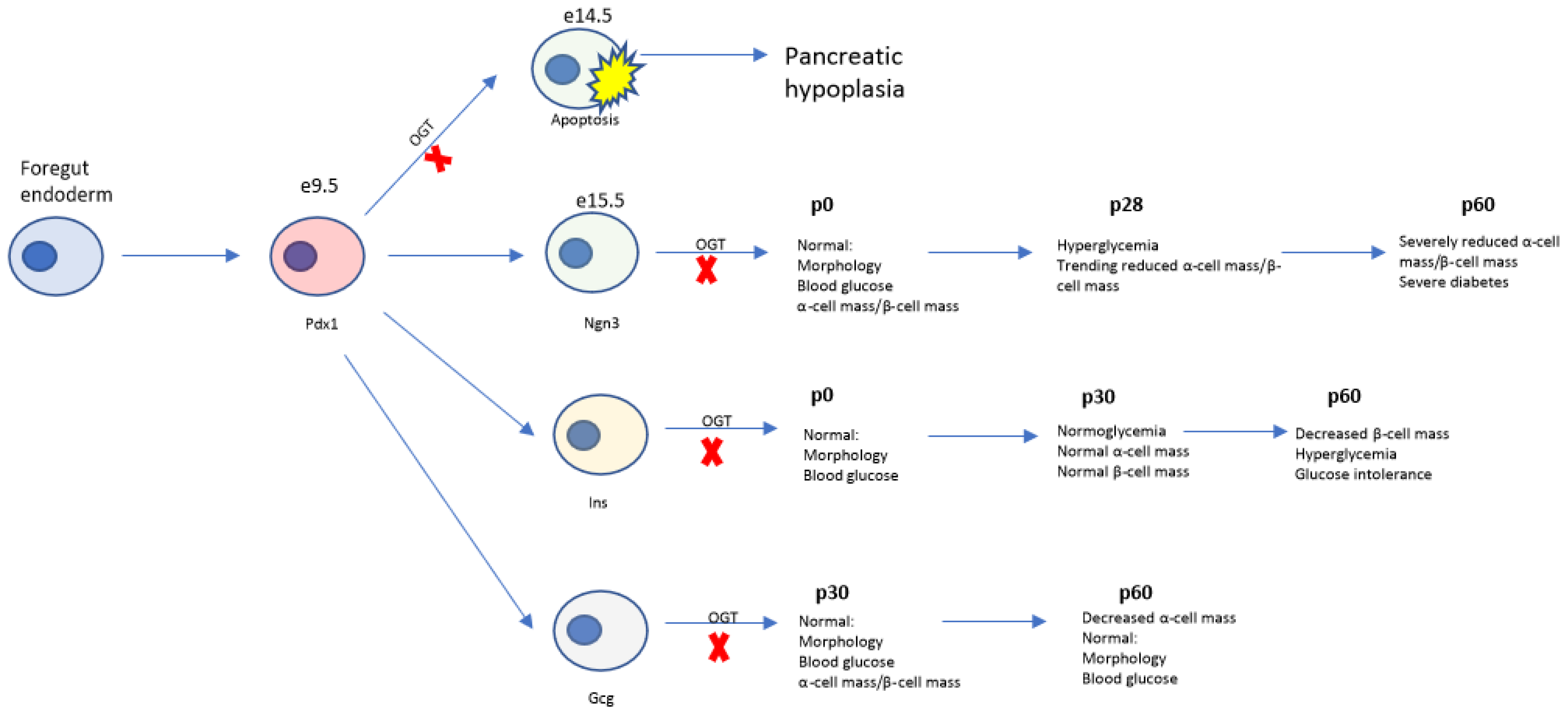
4. Discussion
4.1. Role of Islet Protein O-GlcNAcylation in Glucose Homeostasis
4.2. Maintenance of the α-Cell and β-Cell Population
4.3. Pro-Survival Role of Ogt in the Endocrine Cells
4.4. Temporal Role of Ogt in Regulation of Pancreas Development
4.5. Limitations of the Study
4.6. Speculation on Clinical Utility of Present Data
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef]
- Holt, G.D.; Hart, G.W. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J. Biol. Chem. 1986, 261, 8049–8057. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Yu, S.; Lubas, W.B.; Shin, S.H.; Ragano-Caracciola, M.; Kochran, J.; Love, D.C. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch. Biochem. Biophys. 2003, 409, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Love, D.C.; Hanover, J.A. Elevated O-GlcNAc-dependent signaling through inducible mOGT expression selectively triggers apoptosis. Amino Acids 2011, 40, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, D.M.; Fu, D.J.; Freeman, A.B.; Hunt, K.J.; Leach, R.J.; Johnson-Pais, T.; Hamlington, J.; Dyer, T.D.; Arya, R.; Abboud, H.; et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes 2005, 54, 1214–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaveroux, C.; Sarcinelli, C.; Barbet, V.; Belfeki, S.; Barthelaix, A.; Ferraro-Peyret, C.; Lebecque, S.; Renno, T.; Bruhat, A.; Fafournoux, P.; et al. Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2-ATF4 signalling pathway. Sci. Rep. 2016, 6, 27278. [Google Scholar] [CrossRef] [Green Version]
- Slawson, C.; Housley, M.P.; Hart, G.W. O-GlcNAc cycling: How a single sugar post-translational modification is changing the way we think about signaling networks. J. Cell. Biochem. 2006, 97, 71–83. [Google Scholar] [CrossRef]
- Zachara, N.E.; Hart, G.W. O-GlcNAc a sensor of cellular state: The role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta 2004, 1673, 13–28. [Google Scholar] [CrossRef]
- Dias, W.B.; Hart, G.W. O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol. Biosyst. 2007, 3, 766–772. [Google Scholar] [CrossRef]
- Buse, M.G. Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1–E8. [Google Scholar] [CrossRef]
- Copeland, R.J.; Bullen, J.W.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Roles in insulin resistance and glucose toxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E17–E28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slawson, C.; Copeland, R.J.; Hart, G.W. O-GlcNAc signaling: A metabolic link between diabetes and cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Myslicki, J.P.; Shearer, J.; Hittel, D.S.; Hughey, C.C.; Belke, D.D. O-GlcNAc modification is associated with insulin sensitivity in the whole blood of healthy young adult males. Diabetol. Metab. Syndr. 2014, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubas, W.A.; Frank, D.W.; Krause, M.; Hanover, J.A. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J. Biol. Chem. 1997, 272, 9316–9324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro, E.U.; Bozadjieva, N.; Kumusoglu, D.; Abdulhamid, S.; Levine, H.; Haataja, L.; Vadrevu, S.; Satin, L.S.; Arvan, P.; Bernal-Mizrachi, E. Disruption of O-linked N-Acetylglucosamine Signaling Induces ER Stress and beta Cell Failure. Cell Rep. 2015, 13, 2527–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassaye, R.; Naidoo, S.; Cerf, M.E. Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets 2016, 8, 13–34. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson, H.; Karlsson, K.; Edlund, T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993, 12, 4251–4259. [Google Scholar] [CrossRef]
- Hart, G.W. Nutrient regulation of signaling and transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Miyazaki, J.; Hart, G.W. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch. Biochem. Biophys. 2003, 415, 155–163. [Google Scholar] [CrossRef]
- Baumann, D.; Wong, A.; Akhaphong, B.; Jo, S.; Pritchard, S.; Mohan, R.; Chung, G.; Zhang, Y.; Alejandro, E.U. Role of nutrient-driven O-GlcNAc-post-translational modification in pancreatic exocrine and endocrine islet development. Development 2020, 147, dev186643. [Google Scholar] [CrossRef]
- Lockridge, A.; Jo, S.; Gustafson, E.; Damberg, N.; Mohan, R.; Olson, M.; Abrahante, J.E.; Alejandro, E.U. Islet O-GlcNAcylation Is Required for Lipid Potentiation of Insulin Secretion through SERCA2. Cell Rep. 2020, 31, 107609. [Google Scholar] [CrossRef] [PubMed]
- Lamarre-Vincent, N.; Hsieh-Wilson, L.C. Dynamic glycosylation of the transcription factor CREB: A potential role in gene regulation. J. Am. Chem. Soc. 2003, 125, 6612–6613. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ongusaha, P.P.; Miles, P.D.; Havstad, J.C.; Zhang, F.; So, W.V.; Kudlow, J.E.; Michell, R.H.; Olefsky, J.M.; Field, S.J.; et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008, 451, 964–969. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Pritchard, S.; Wong, A.; Avula, N.; Essawy, A.; Hanover, J.; Alejandro, E.U. Pancreatic β-cell hyper-O-GlcNAcylation leads to impaired glucose homeostasis in vivo. Front. Endocrinol. 2022, 13, 1040014. [Google Scholar] [CrossRef]
- Akhaphong, B.; Lockridge, A.; Jo, S.; Mohan, R.; Wilcox, J.A.; Wing, C.R.; Regal, J.F.; Alejandro, E.U. Reduced uterine perfusion pressure causes loss of pancreatic β-cell area but normal function in fetal rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1220–R1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, L.; Cahill, C.J.; Finegood, D.T.; Bonner-Weir, S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997, 138, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Mohan, R.; Jo, S.; Da Sol Chung, E.; Oribamise, E.; Lockridge, A.; Abrahante-Lloréns, J.E.; Ruan, H.B.; Yang, X.Y.; Alejandro, E.U. Pancreatic β-Cell O-GlcNAc Transferase Overexpression Increases Susceptibility to Metabolic Stressors in Female Mice. Cells 2021, 10, 2801. [Google Scholar] [CrossRef]
- Essawy, A.; Jo, S.; Beetch, M.; Lockridge, A.; Gustafson, E.; Alejandro, E.U. O-linked N-acetylglucosamine transferase (OGT) regulates pancreatic α-cell function in mice. J. Biol. Chem. 2021, 296, 100297. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Diaz, R.; Molano, R.D.; Weitz, J.R.; Abdulreda, M.H.; Berman, D.M.; Leibiger, B.; Leibiger, I.B.; Kenyon, N.S.; Ricordi, C.; Pileggi, A.; et al. Paracrine Interactions within the Pancreatic Islet Determine the Glycemic Set Point. Cell Metab. 2018, 27, 549–558.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamori, D.; Kurpad, A.J.; Hu, J.; Liew, C.W.; Shih, J.L.; Ford, E.L.; Herrera, P.L.; Polonsky, K.S.; McGuinness, O.P.; Kulkarni, R.N. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab. 2009, 9, 350–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huypens, P.; Ling, Z.; Pipeleers, D.; Schuit, F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 2000, 43, 1012–1019. [Google Scholar] [CrossRef] [Green Version]
- Herrera, P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000, 127, 2317–2322. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, Q.; Zhou, Z.; Ikeda, Y. PDX1, Neurogenin-3, and MAFA: Critical transcription regulators for beta cell development and regeneration. Stem Cell Res. Ther. 2017, 8, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D.; Ahmed, N.T.; Luciani, D.S.; Han, Z.; Tran, H.; Fujita, J.; Misler, S.; Edlund, H.; Polonsky, K.S. Increased islet apoptosis in Pdx1+/− mice. J. Clin. Investig. 2003, 111, 1147–1160. [Google Scholar] [CrossRef]
- Wong, A.; Pritchard, S..; Moore, M.; Akhaphong, B.; Avula, N.; Beetch, M.; Fujitani, Y.; Alejandro, E.U. Overexpression of Pdx1, reduction of p53, or deletion of CHOP attenuates pancreas hypoplasia in mice with pancreas-specific O-GlcNAc Transferase deletion. J. Biol. Chem. 2022. [Google Scholar]
- Gannon, M.; Ables, E.T.; Crawford, L.; Lowe, D.; Offield, M.F.; Magnuson, M.A.; Wright, C.V. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev. Biol. 2008, 314, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Wicksteed, B.; Brissova, M.; Yan, W.; Opland, D.M.; Plank, J.L.; Reinert, R.B.; Dickson, L.M.; Tamarina, N.A.; Philipson, L.H.; Shostak, A.; et al. Conditional gene targeting in mouse pancreatic ß-Cells: Analysis of ectopic Cre transgene expression in the brain. Diabetes 2010, 59, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Lagerlof, O.; Slocomb, J.E.; Hong, I.; Aponte, Y.; Blackshaw, S.; Hart, G.W.; Huganir, R.L. The nutrient sensor OGT in PVN neurons regulates feeding. Science 2016, 351, 1293–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Xiong, X.; Ren, K.; Xu, B.; Cheng, M.; Sahu, C.; Wu, K.; Nie, Y.; Huang, Z.; Blumberg, R.S.; et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol. Med. 2018, 10, e8736. [Google Scholar] [CrossRef]
- Grarup, N.; Sparsø, T.; Hansen, T. Physiologic characterization of type 2 diabetes-related loci. Curr. Diab. Rep. 2010, 10, 485–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnurr, T.M.; Jakupović, H.; Carrasquilla, G.D.; Ängquist, L.; Grarup, N.; Sørensen, T.I.A.; Tjønneland, A.; Overvad, K.; Pedersen, O.; Hansen, T.; et al. Obesity, unfavourable lifestyle and genetic risk of type 2 diabetes: A case-cohort study. Diabetologia 2020, 63, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, A.; Akhaphong, B.; Baumann, D.; Alejandro, E.U. Genetic Ablation of the Nutrient Sensor Ogt in Endocrine Progenitors Is Dispensable for β-Cell Development but Essential for Maintenance of β-Cell Mass. Biomedicines 2023, 11, 105. https://doi.org/10.3390/biomedicines11010105
Wong A, Akhaphong B, Baumann D, Alejandro EU. Genetic Ablation of the Nutrient Sensor Ogt in Endocrine Progenitors Is Dispensable for β-Cell Development but Essential for Maintenance of β-Cell Mass. Biomedicines. 2023; 11(1):105. https://doi.org/10.3390/biomedicines11010105
Chicago/Turabian StyleWong, Alicia, Brian Akhaphong, Daniel Baumann, and Emilyn U. Alejandro. 2023. "Genetic Ablation of the Nutrient Sensor Ogt in Endocrine Progenitors Is Dispensable for β-Cell Development but Essential for Maintenance of β-Cell Mass" Biomedicines 11, no. 1: 105. https://doi.org/10.3390/biomedicines11010105
APA StyleWong, A., Akhaphong, B., Baumann, D., & Alejandro, E. U. (2023). Genetic Ablation of the Nutrient Sensor Ogt in Endocrine Progenitors Is Dispensable for β-Cell Development but Essential for Maintenance of β-Cell Mass. Biomedicines, 11(1), 105. https://doi.org/10.3390/biomedicines11010105