
A Novel Frameshift CHD4 Variant Leading to Sifrim-Hitz-Weiss Syndrome in a Proband with a Subclinical Familial t(17;19) and a Large dup(2)(q14.3q21.1)
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Karyotyping, Genomic DNA Extraction and Sanger Sequencing
2.2. Genome and Exome Sequencing
2.3. Sequencing Data Analysis and Variant Interpretation
3. Case Report
3.1. Clinical Description
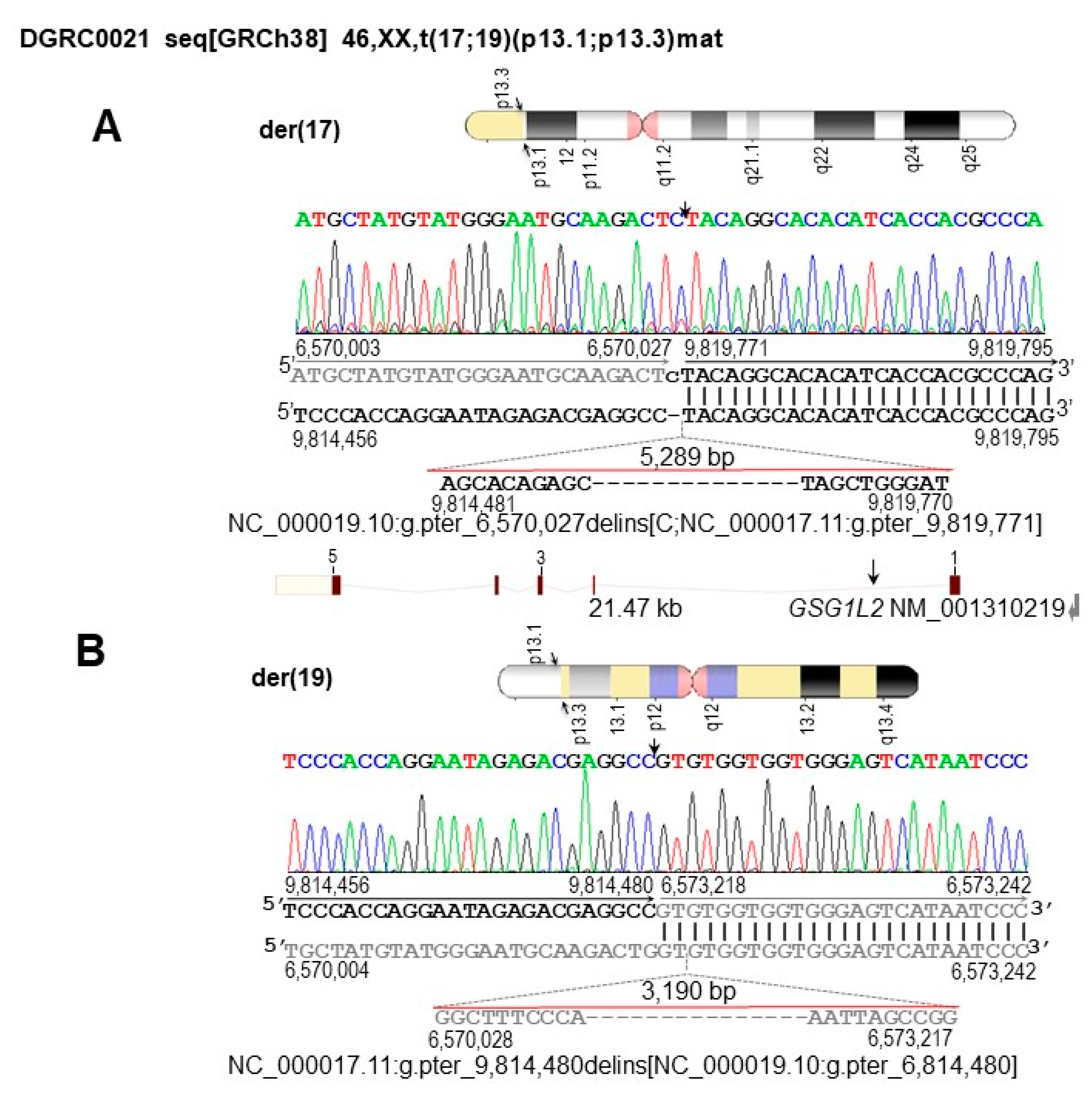
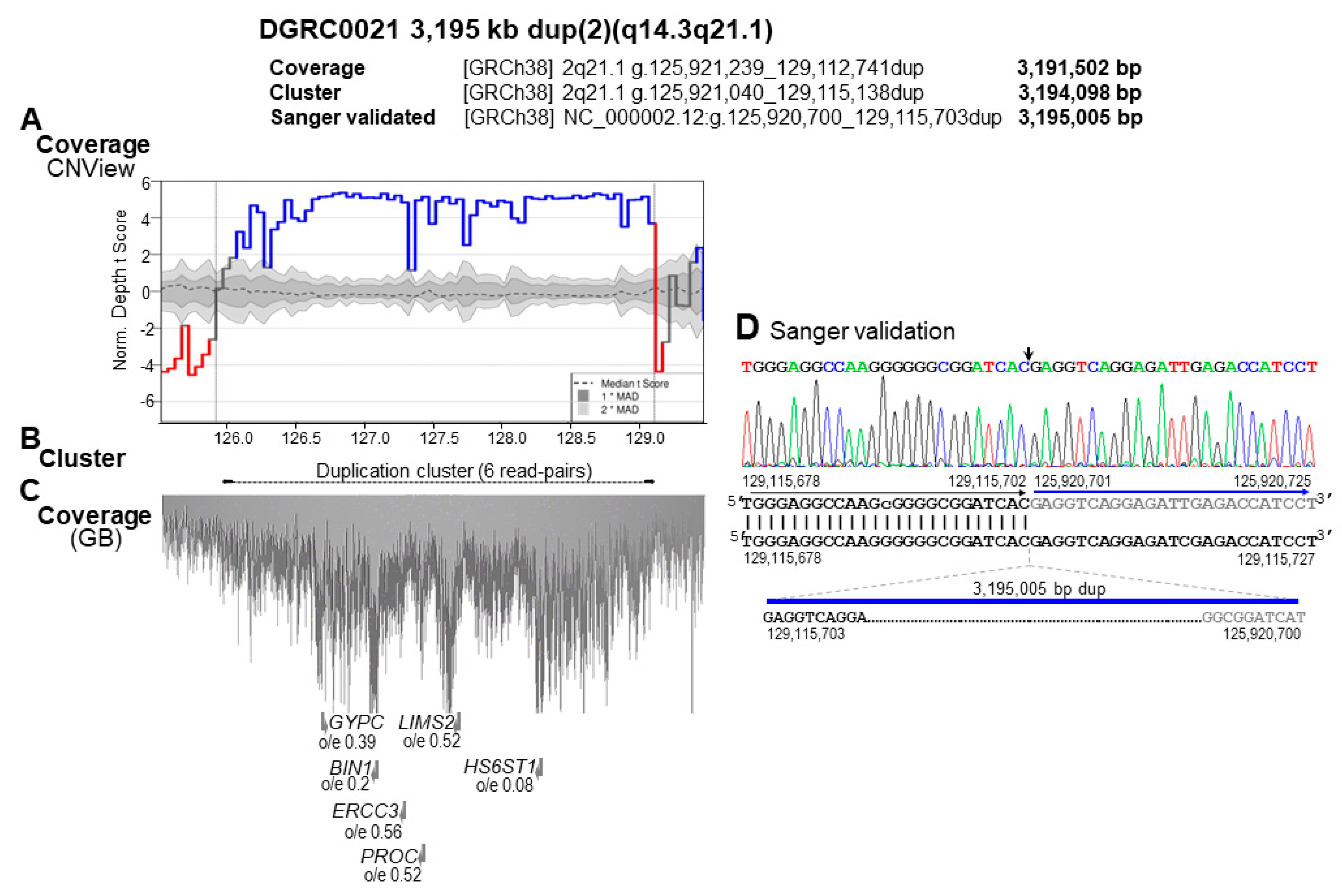
3.2. Identification of SV and CNV Breakpoints at Nucleotide Resolution
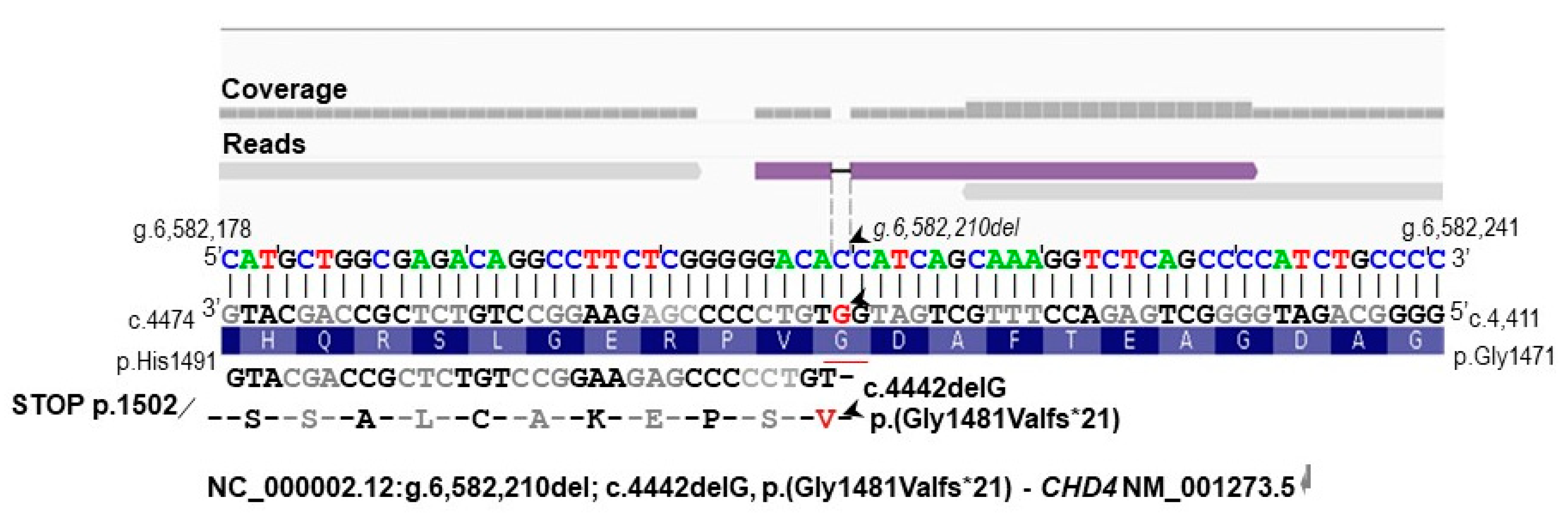
3.3. Identification of Additional Genetic Defects
3.4. Characterization of the Genomic and Genetic Variants and Affected Candidate Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Redin, C.; Brand, H.; Collins, R.L.; Kammin, T.; Mitchell, E.; Hodge, J.C.; Hanscom, C.; Pillalamarri, V.; Seabra, C.M.; Abbott, M.-A.; et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017, 49, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, D.; Freixo, J.P.; Fino, J.; Carvalho, I.; Marques, M.; Cardoso, M.; Piña-Aguilar, R.E.; Morton, C.C. Comprehensive clinically oriented workflow for nucleotide level resolution and interpretation in prenatal diagnosis of de novo apparently balanced chromosomal translocations in their genomic landscape. Hum. Genet. 2020, 139, 531–543. [Google Scholar] [CrossRef]
- Köhler, S.; Carmody, L.; Vasilevsky, N.; Jacobsen, J.O.B.; Danis, D.; Gourdine, J.-P.; Gargano, M.; Harris, N.L.; Matentzoglu, N.; McMurry, J.A.; et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019, 47, D1018–D1027. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Gao, L.; Wang, B.; Guo, X. HPOSim: An R package for phenotypic similarity measure and enrichment analysis based on the human phenotype ontology. PLoS ONE 2015, 10, e0115692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fino, J.; Marques, B.; Dong, Z.; David, D. SVInterpreter: A Comprehensive Topologically Associated Domain-Based Clinical Outcome Prediction Tool for Balanced and Unbalanced Structural Variants. Front. Genet. 2021, 12, 757170. [Google Scholar] [CrossRef]
- Talkowski, M.E.; Ernst, C.; Heilbut, A.; Chiang, C.; Hanscom, C.; Lindgren, A.; Kirby, A.; Liu, S.; Muddukrishna, B.; Ohsumi, T.K.; et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am. J. Hum. Genet. 2011, 88, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Howe, B.; Umrigar, A.; Tsien, F. Chromosome preparation from cultured cells. J. Vis. Exp. 2014, 83, e50203. [Google Scholar] [CrossRef] [Green Version]
- Guha, P.; Das, A.; Dutta, S.; Chaudhuri, T.K. A rapid and efficient DNA extraction protocol from fresh and frozen human blood samples. J. Clin. Lab. Anal. 2018, 32, e22181. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.-A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S. cn.MOPS: Mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.L.; Stone, M.R.; Brand, H.; Glessner, J.T.; Talkowski, M.E. CNView: A visualization and annotation tool for copy number variation from whole-genome sequencing. Biorxiv 2016, 049536. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.M.; Lawrence, C.E.; Raphael, B.J. Detecting non-allelic homologous recombination from high-throughput sequencing data. Genome Biol. 2015, 16, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Jung, I.; Selvaraj, S.; Shen, Y.; Antosiewicz-Bourget, J.E.; Lee, A.Y.; Ye, Z.; Kim, A.; Rajagopal, N.; Xie, W.; et al. Chromatin architecture reorganization during stem cell differentiation. Nature 2015, 518, 331–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumkin, L.; Halevy, A.; Ben-Ami-Raichman, D.; Dahari, D.; Haviv, A.; Sarit, C.; Lev, D.; van der Knaap, M.S.; Lerman-Sagie, T.; Leshinsky-Silver, E. Expansion of the spectrum of TUBB4A-related disorders: A new phenotype associated with a novel mutation in the TUBB4A gene. Neurogenetics 2014, 15, 107–113. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Terhal, P.A.; Cohen, L.; Bruccoleri, M.; Irving, M.; Martinez, A.F.; Rosenfeld, J.A.; Machol, K.; Yang, Y.; Liu, P.; et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016, 99, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Lachlan, K. CHD4 Neurodevelopmental Disorder; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-related syndrome: A comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef]
- Pinard, A.; Guey, S.; Guo, D.; Cecchi, A.C.; Kharas, N.; Wallace, S.; Regalado, E.S.; Hostetler, E.M.; Sharrief, A.Z.; Bergametti, F.; et al. The pleiotropy associated with de novo variants in CHD4, CNOT3, and SETD5 extends to moyamoya angiopathy. Genet. Med. 2020, 22, 427–431. [Google Scholar] [CrossRef]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018, 10, 56. [Google Scholar] [CrossRef]
- Brunet, T.; Jech, R.; Brugger, M.; Kovacs, R.; Alhaddad, B.; Leszinski, G.; Riedhammer, K.M.; Westphal, D.S.; Mahle, I.; Mayerhanser, K.; et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin. Genet. 2021, 100, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-R.; Ye, T.-T.; Zhang, W.-J.; Guo, X.; Wang, J.; Huang, S.-P.; Xie, L.-S.; Song, X.-W.; Deng, W.-W.; Li, B.-M.; et al. CHD4 variants are associated with childhood idiopathic epilepsy with sinus arrhythmia. CNS Neurosci. Ther. 2021, 27, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Q.; Liu, Y.; Liu, J.; Yuan, H. Clinical and genetic analysis of a novel CHD4 gene variant in a Chinese patient with Sifrim-Hitz-Weiss syndrome. Chin. J. Med. Genet. 2021, 38, 63–66. [Google Scholar] [CrossRef]
- Dahlbäck, B.; Villoutreix, B.O. The anticoagulant protein C pathway. FEBS Lett. 2005, 579, 3310–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouidja, M.O.; Biard, D.S.F.; Chantepie, S.; Laffray, X.; Le Douaron, G.; Huynh, M.-B.; Rebergue, N.; Maïza, A.; Rubio, K.; González-Velasco, O. Variability in proteoglycan biosynthetic genes reveals new facets of heparan sulfates diversity. A systematic review and analysis. Medrxiv 2022. [Google Scholar] [CrossRef]
- Ihara, M.; Yamamoto, Y.; Hattori, Y.; Liu, W.; Kobayashi, H.; Ishiyama, H.; Yoshimoto, T.; Miyawaki, S.; Clausen, T.; Bang, O.Y.; et al. Moyamoya disease: Diagnosis and interventions. Lancet Neurol. 2022, 21, 747–758. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Proband’s Clinical Features, HPO Term | SIHIWES or CHD4-NDDpatients | ||||
|---|---|---|---|---|---|
| OMIM #617159 | All Variants (%) | Missense Variants (%) | Truncating Variants (%) | Splicing Variants | |
| Growth | |||||
| Obesity, HP:0001513 | No | 3/36 (8.3) | 3/31 (9.7) | 0/3 (0.0) | 0/2 |
| Head and neck | |||||
| Coarse facies, HP:0000280 | Yes | 0/44 (0.0) | 0/38 (0.0) | 0/4 (0.0) | 0/2 |
| Muscoloskeletal | |||||
| Hypotonia, HP:0001252 | Yes | 18/35 (51.4) | 16/30 (53.3) | 2/3 (66.7) | 0/2 |
| Macrocephaly, HP:0000256 | Yes | 13/40 (32.5) | 11/35 (31.4) | 2/3 (66.7) | 0/2 |
| Nervous system | |||||
| Agitation, HP:0000713 | No | 0/44 (0.0) | 0/38 (0.0) | 0/4 (0.0) | 0/2 |
| Aggressive behavior, HP:0000718 | No | 0/44 (0.0) | 0/38 (0.0) | 0/4 (0.0) | 0/2 |
| Anxiety. HP:0000739 | No | 2/36 (5.6) | 2/31 (6.5) | 0/4 (0.0) | 0/2 |
| Emotional lability, HP:0000712 | No | 0/44 (0.0) | 0/38 (0.0) | 0/4 (0.0) | 0/2 |
| Global developmental delay, HP:0001263 | Yes | 2/44 (4.6) | 2/38 (5.3) | 0/4 (0.0) | 0/2 |
| Headache, HP:0002315 | No | 2/44 (4.5) | 2/38 (5.3) | 0/4 (0.0) | 0/2 |
| Hydrocephalus, HP:0000238 | No | 5/44 (11.4) | 5/38 (13.2) | 0/4 (0.0) | 0/2 |
| Intellectual disability, HP:0001249 | Yes | 20/34 (58.8) | 19/31 (61.3) | 1/1 (100) | 0/2 |
| Motor delay, HP:0001270 | No | 29/43 (67.4) | 27/38 (71.1) | 2/3 (66.7) | 0/2 |
| Speech delay, HP:0000750 | No | 31/43 (72.1) | 28/38 (73.8) | 3/3 (100) | 0/2 |
| Phenotype similarity score a PhenSSc (p-value) | 1.29 (0.20) | 2.33 (0.06) | 1.21 (0.07) | 1.22 (0.01) | 0.81 (0.16) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Da Silva, J.D.; Oliva-Teles, N.; Tkachenko, N.; Fino, J.; Marques, M.; Fortuna, A.M.; David, D. A Novel Frameshift CHD4 Variant Leading to Sifrim-Hitz-Weiss Syndrome in a Proband with a Subclinical Familial t(17;19) and a Large dup(2)(q14.3q21.1). Biomedicines 2023, 11, 12. https://doi.org/10.3390/biomedicines11010012
Da Silva JD, Oliva-Teles N, Tkachenko N, Fino J, Marques M, Fortuna AM, David D. A Novel Frameshift CHD4 Variant Leading to Sifrim-Hitz-Weiss Syndrome in a Proband with a Subclinical Familial t(17;19) and a Large dup(2)(q14.3q21.1). Biomedicines. 2023; 11(1):12. https://doi.org/10.3390/biomedicines11010012
Chicago/Turabian StyleDa Silva, Jorge Diogo, Natália Oliva-Teles, Nataliya Tkachenko, Joana Fino, Mariana Marques, Ana Maria Fortuna, and Dezso David. 2023. "A Novel Frameshift CHD4 Variant Leading to Sifrim-Hitz-Weiss Syndrome in a Proband with a Subclinical Familial t(17;19) and a Large dup(2)(q14.3q21.1)" Biomedicines 11, no. 1: 12. https://doi.org/10.3390/biomedicines11010012
APA StyleDa Silva, J. D., Oliva-Teles, N., Tkachenko, N., Fino, J., Marques, M., Fortuna, A. M., & David, D. (2023). A Novel Frameshift CHD4 Variant Leading to Sifrim-Hitz-Weiss Syndrome in a Proband with a Subclinical Familial t(17;19) and a Large dup(2)(q14.3q21.1). Biomedicines, 11(1), 12. https://doi.org/10.3390/biomedicines11010012