
Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Breeding
2.2. Ganglioside Analysis
2.3. Motor and Memory Impairment Tests
2.4. Statistical Analysis
3. Results
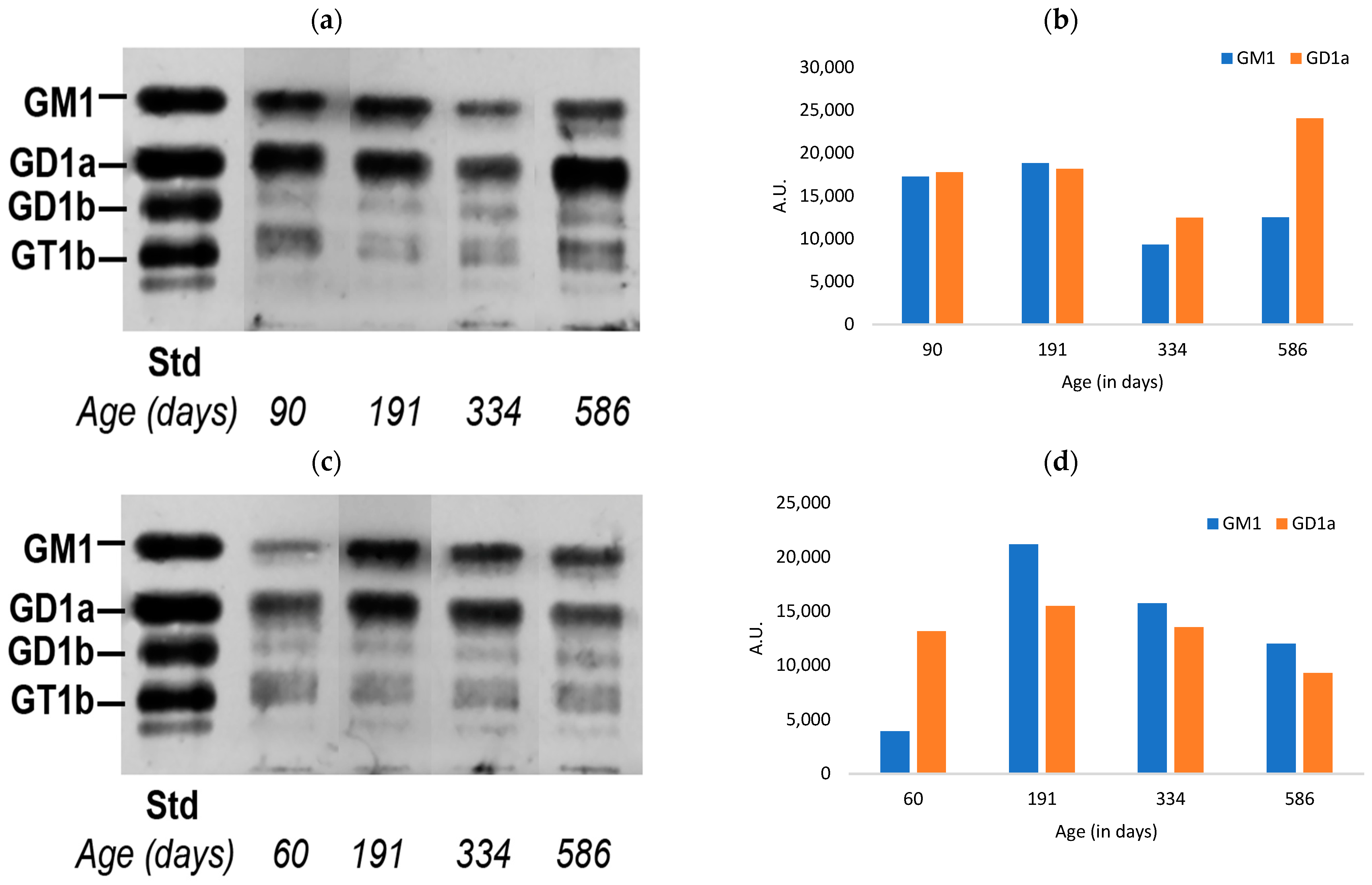
3.1. Ganglioside Changes in Peripheral Tissues of Normal Mice with Aging
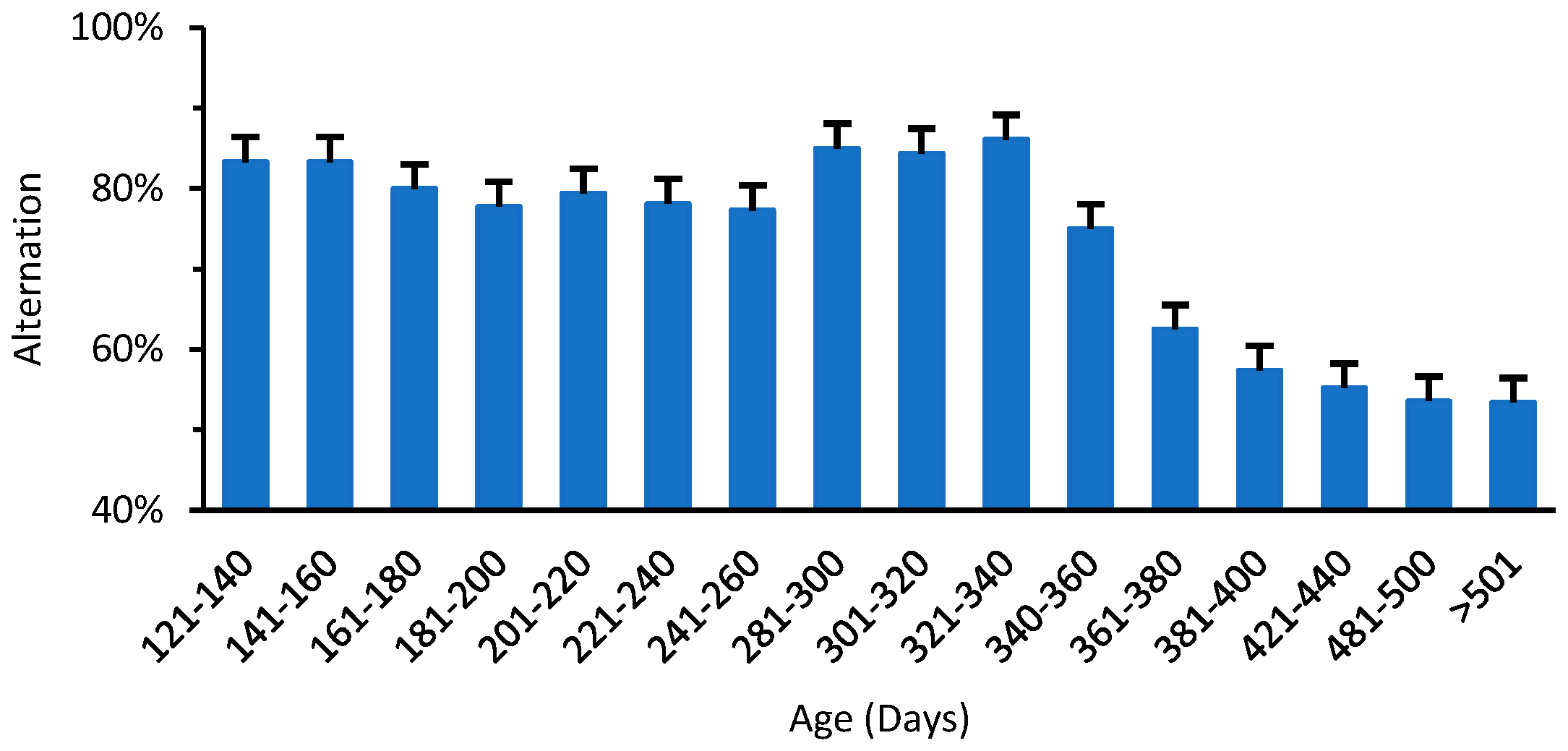
3.2. Changes in Motor Function of Normal Mice with Aging
3.3. Changes in the Memory Impairment of Normal Mice with Aging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galpern, W.R.; Lang, A.E. Interface between tauopathies and synucleinopathies: A tale of two proteins. Ann. Neurol. 2006, 59, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; de Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Braak, H.; Bohl, J.R.; Müller, C.M.; Rüb, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 2042–2051. [Google Scholar] [CrossRef]
- Pellicano, C.; Benincasa, D.; Pisani, V.; Buttarelli, F.R.; Giovannelli, M.; Pontieri, F.E. Prodromal non-motor symptoms of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Borghammer, P. How does Parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov. Disord. 2018, 33, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641.e627. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Van Den Berge, N. Brain-first versus gut-first Parkinson’s disease: A hypothesis. J. Park. Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef] [Green Version]
- Rietdijk, C.D.; Perez-Pardo, P.; Garssen, J.; Van Wezel, R.J.; Kraneveld, A.D. Exploring Braak’s hypothesis of Parkinson’s disease. Front. Neurol. 2017, 8, 37. [Google Scholar] [CrossRef]
- Parkkinen, L.; Pirttilä, T.; Alafuzoff, I. Applicability of current staging/categorization of α-synuclein pathology and their clinical relevance. Acta Neuropathol. 2008, 115, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raunio, A.; Kaivola, K.; Tuimala, J.; Kero, M.; Oinas, M.; Polvikoski, T.; Paetau, A.; Tienari, P.J.; Myllykangas, L. Lewy-related pathology exhibits two anatomically and genetically distinct progression patterns: A population-based study of Finns aged 85+. Acta Neuropathol. 2019, 138, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef]
- Ledeen, R.; Chowdhury, S.; Lu, Z.-H.; Chakraborty, M.; Wu, G. Systemic deficiency of GM1 ganglioside in Parkinson’s disease tissues and its relation to the disease etiology. Glycoconj. J. 2022, 39, 75–82. [Google Scholar] [CrossRef]
- Uchihara, T.; Giasson, B.I. Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 49–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Lu, Z.-H.; Gabius, H.-J.; Ledeen, R.W.; Bleich, D. Ganglioside GM1 deficiency in effector T cells from NOD mice induces resistance to regulatory T-cell suppression. Diabetes 2011, 60, 2341–2349. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Ledeen, R. Quantification of gangliotetraose gangliosides with cholera toxin. Anal. Biochem. 1988, 173, 368–375. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.-H.; Seo, J.H.; Alselehdar, S.K.; DeFrees, S.; Ledeen, R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020, 329, 113284. [Google Scholar] [CrossRef]
- Hadaczek, P.; Wu, G.; Sharma, N.; Ciesielska, A.; Bankiewicz, K.; Davidow, A.L.; Lu, Z.-H.; Forsayeth, J.; Ledeen, R.W. GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp. Neurol. 2015, 263, 177–189. [Google Scholar] [CrossRef]
- Fazzari, M.; Di Biase, E.; Lunghi, G.; Mauri, L.; Chiricozzi, E.; Sonnino, S. Novel insights on GM1 and Parkinson’s disease: A critical review. Glycoconj. J. 2022, 39, 27–38. [Google Scholar] [CrossRef]
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Boström, K.; Fredman, P.; Månsson, J.-E.; Rosengren, B.; Rynmark, B.-M. Human brain gangliosides: Developmental changes from early fetal stage to advanced age. Biochim. Et Biophys. Acta BBA Lipids Lipid Metab. 1989, 1005, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Ledeen, R. The Key Role of GM1 Ganglioside in Parkinson’s Disease. Biomolecules 2022, 12, 173. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.; Miller, S. Parkinson’s disease and the skin. Pract. Neurol. 2015, 15, 246–249. [Google Scholar] [CrossRef]
- Goldstein, D.S. Dysautonomia in Parkinson’s disease: Neurocardiological abnormalities. Lancet Neurol. 2003, 2, 669–676. [Google Scholar] [CrossRef]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J. Sex-related abnormalities in substantia nigra lipids in Parkinson’s disease. ASN Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, T.; Yamaguchi, K. Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896. [Google Scholar] [CrossRef]
- Navarro-Romero, A.; Montpeyo, M.; Martinez-Vicente, M. The emerging role of the lysosome in Parkinson’s disease. Cells 2020, 9, 2399. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 2015, 40, 407–418. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with α-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 ganglioside modifies α-synuclein toxicity and is neuroprotective in a rat α-synuclein model of Parkinson’s disease. Sci. Rep. 2019, 9, 8362. [Google Scholar] [CrossRef] [Green Version]
- Boukhris, A.; Schule, R.; Loureiro, J.L.; Lourenço, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Lebrigio, R.F.A. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racette, B.A.; Good, L.M.; Kissel, A.M.; Criswell, S.R.; Perlmutter, J.S. A population-based study of parkinsonism in an Amish community. Neuroepidemiology 2009, 33, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Pascual, A.; Hidalgo-Figueroa, M.; Piruat, J.I.; Pintado, C.O.; Gomez-Diaz, R.; López-Barneo, J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 2008, 11, 755–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counts, S.E.; Nadeem, M.; Wuu, J.; Ginsberg, S.D.; Saragovi, H.U.; Mufson, E.J. Reduction of cortical TrkA but not p75NTR protein in early-stage Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 56, 520–531. [Google Scholar] [CrossRef]
- Guo, Y.L.; Duan, W.J.; Lu, D.H.; Ma, X.H.; Li, X.X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.Z.; Li, Y.F.; et al. Autophagy-dependent removal of α-synuclein: A novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol. Sin. 2021, 42, 518–528. [Google Scholar] [CrossRef]
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 ganglioside in Parkinson’s disease: Results of a five-year open study. J. Neurol. Sci. 2010, 292, 45–51. [Google Scholar] [CrossRef]
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.S. A critical role for GM1 ganglioside in the pathophysiology and potential treatment of Parkinson’s disease. Glycoconj. J. 2022, 39, 13–26. [Google Scholar] [CrossRef]
- Ledeen, R.; Chowdhury, S. Gangliosides in Neurodegenerative Diseases. In Glycobiology of the Nervous System Advances in Neurobiology; Schengrund, C.L., Yu, R.K., Eds.; Springer: Cham, Switzerland, 2023; Volume 29, pp. 391–418. [Google Scholar]
- Paciotti, S.; Elisabetta, A.; Lucilla, P.; Tommaso, B. Lysosomal ceramide metabolism disorders: Implications in Parkinson’s disease. J. Clin. Med. 2020, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 435–454. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhury, S.; Wu, G.; Lu, Z.-H.; Kumar, R.; Ledeen, R. Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease. Biomedicines 2023, 11, 209. https://doi.org/10.3390/biomedicines11010209
Chowdhury S, Wu G, Lu Z-H, Kumar R, Ledeen R. Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease. Biomedicines. 2023; 11(1):209. https://doi.org/10.3390/biomedicines11010209
Chicago/Turabian StyleChowdhury, Suman, Gusheng Wu, Zi-Hua Lu, Ranjeet Kumar, and Robert Ledeen. 2023. "Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease" Biomedicines 11, no. 1: 209. https://doi.org/10.3390/biomedicines11010209
APA StyleChowdhury, S., Wu, G., Lu, Z.-H., Kumar, R., & Ledeen, R. (2023). Age-Related Decline in Gangliosides GM1 and GD1a in Non-CNS Tissues of Normal Mice: Implications for Peripheral Symptoms of Parkinson’s Disease. Biomedicines, 11(1), 209. https://doi.org/10.3390/biomedicines11010209