

CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model

 ,
,  , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antiproliferative Assay
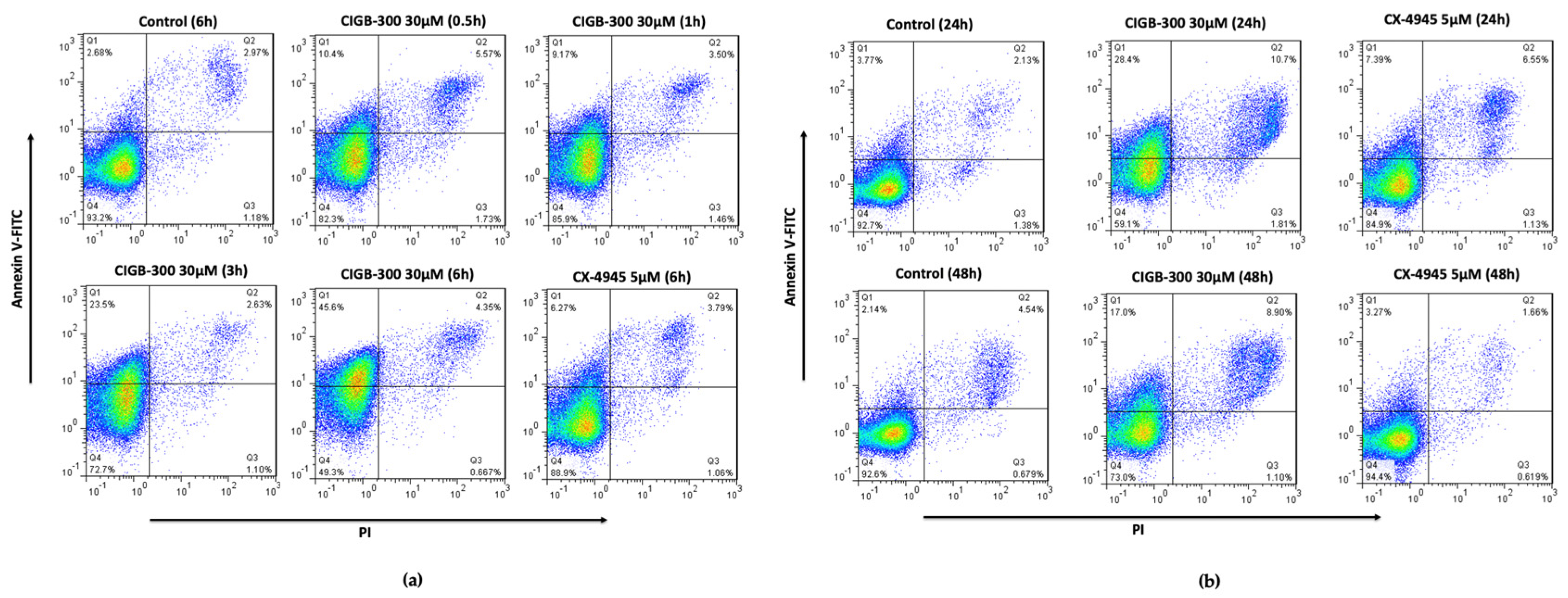
2.3. Annexin V/PI Staining
2.4. Cell Cycle Analysis
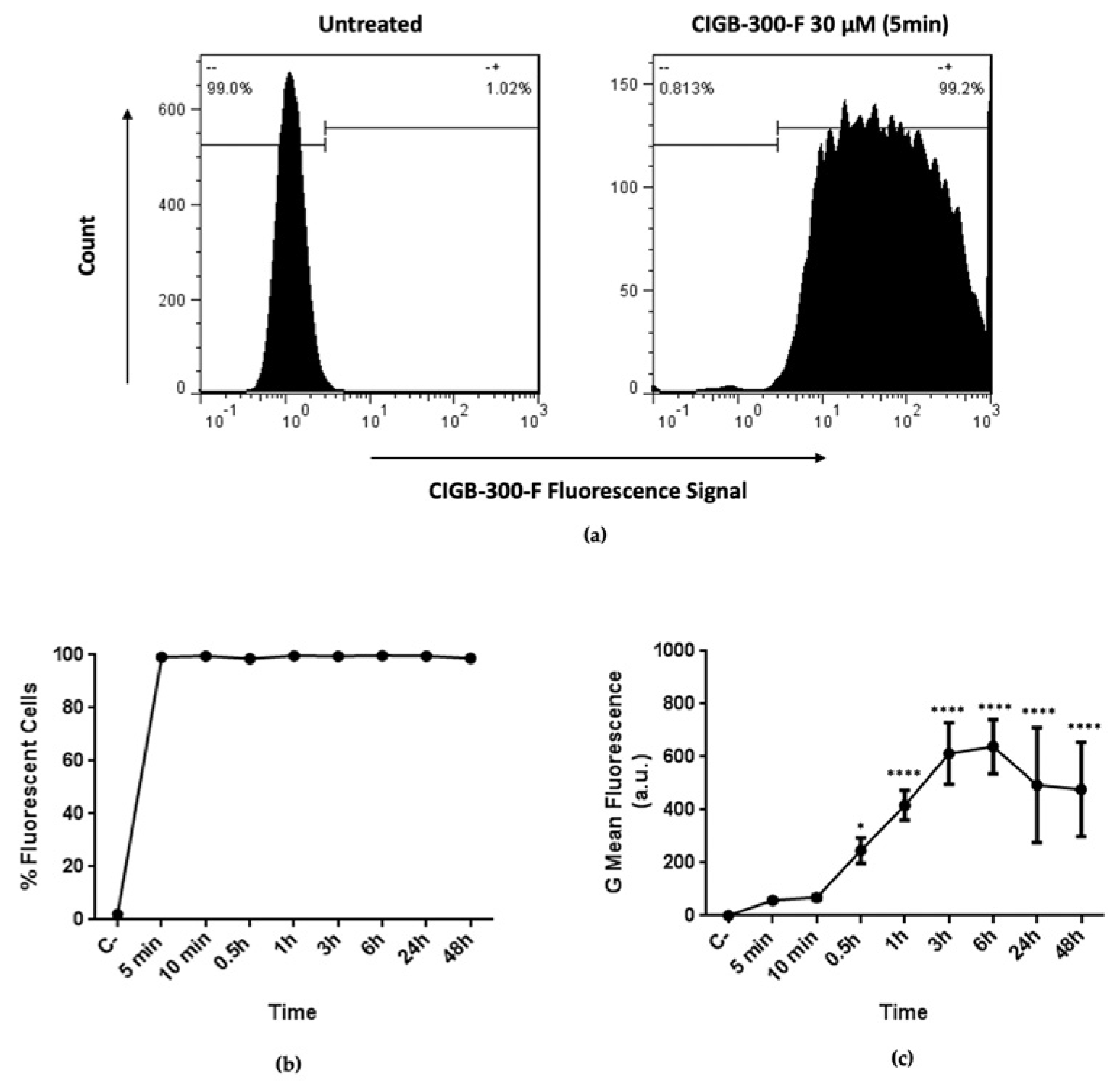
2.5. Peptide Internalization
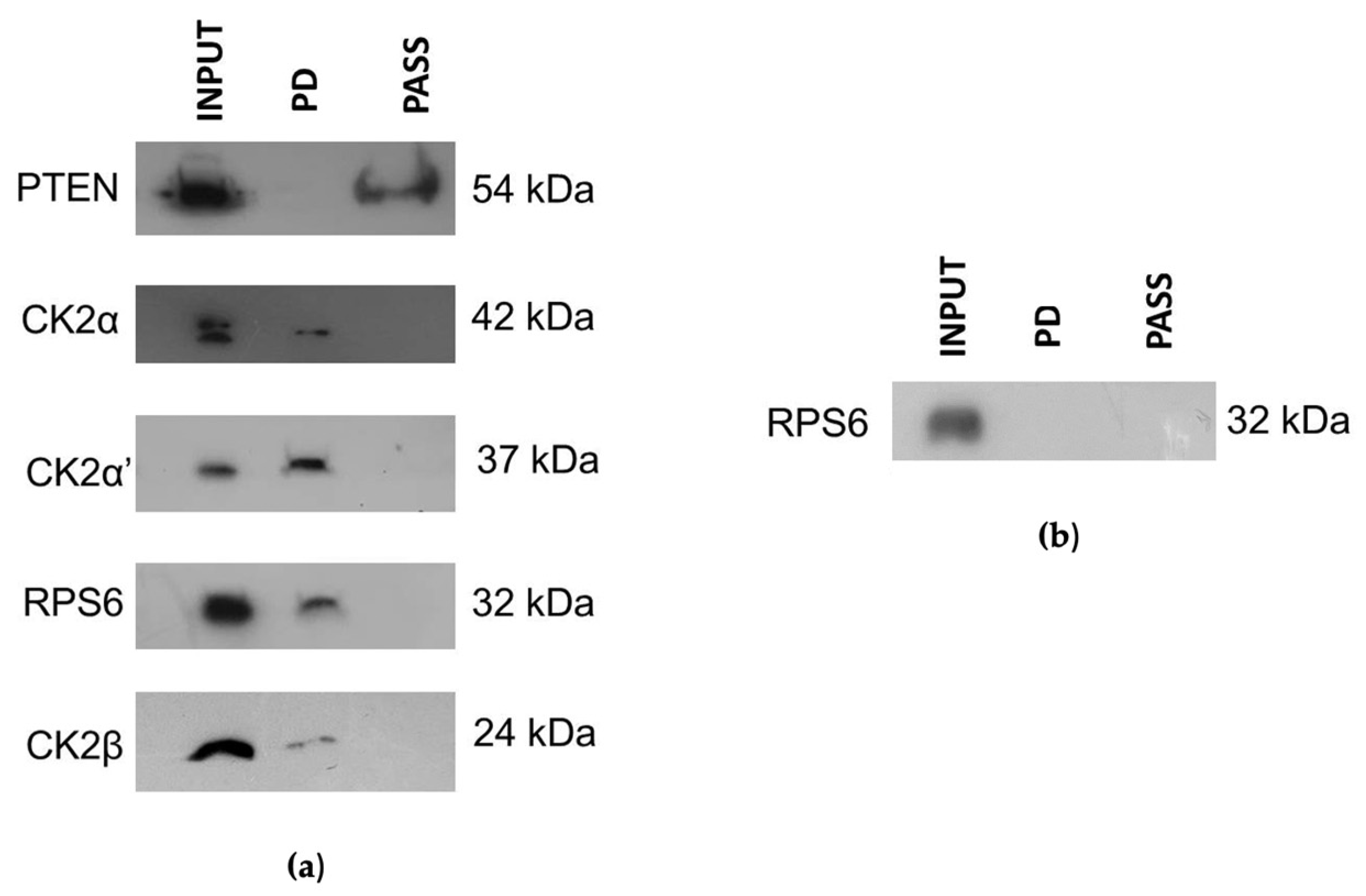
2.6. Pull-Down Experiments
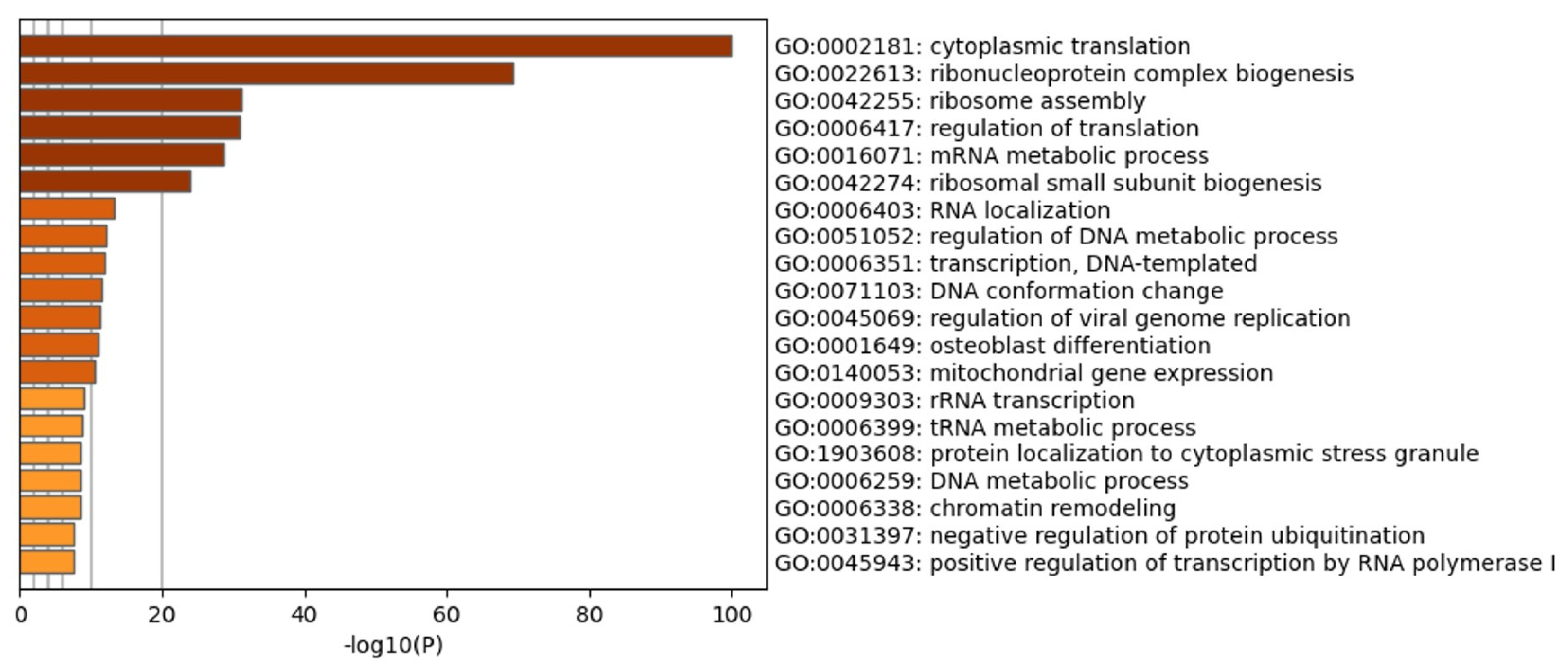
2.7. LC-MS/MS Analysis and Protein Identification
2.8. Confocal Microscopy
2.9. CK2 Subunits and Downstream Signaling
2.10. Quantitative Real-Time PCR Assays
2.11. Statistical Analysis
3. Results
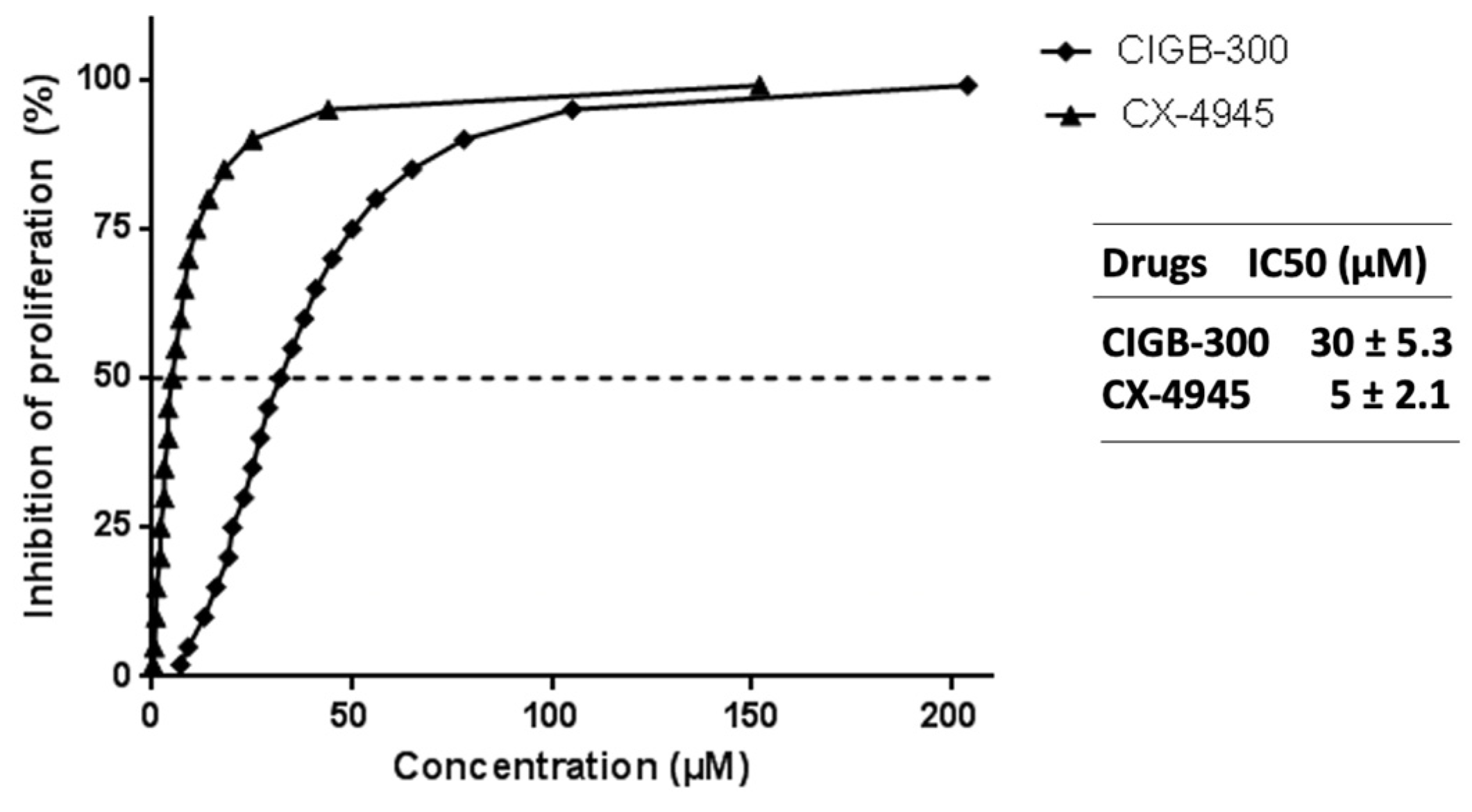
3.1. NCI-H460 Is Highly Sensitive to CIGB-300 Peptide
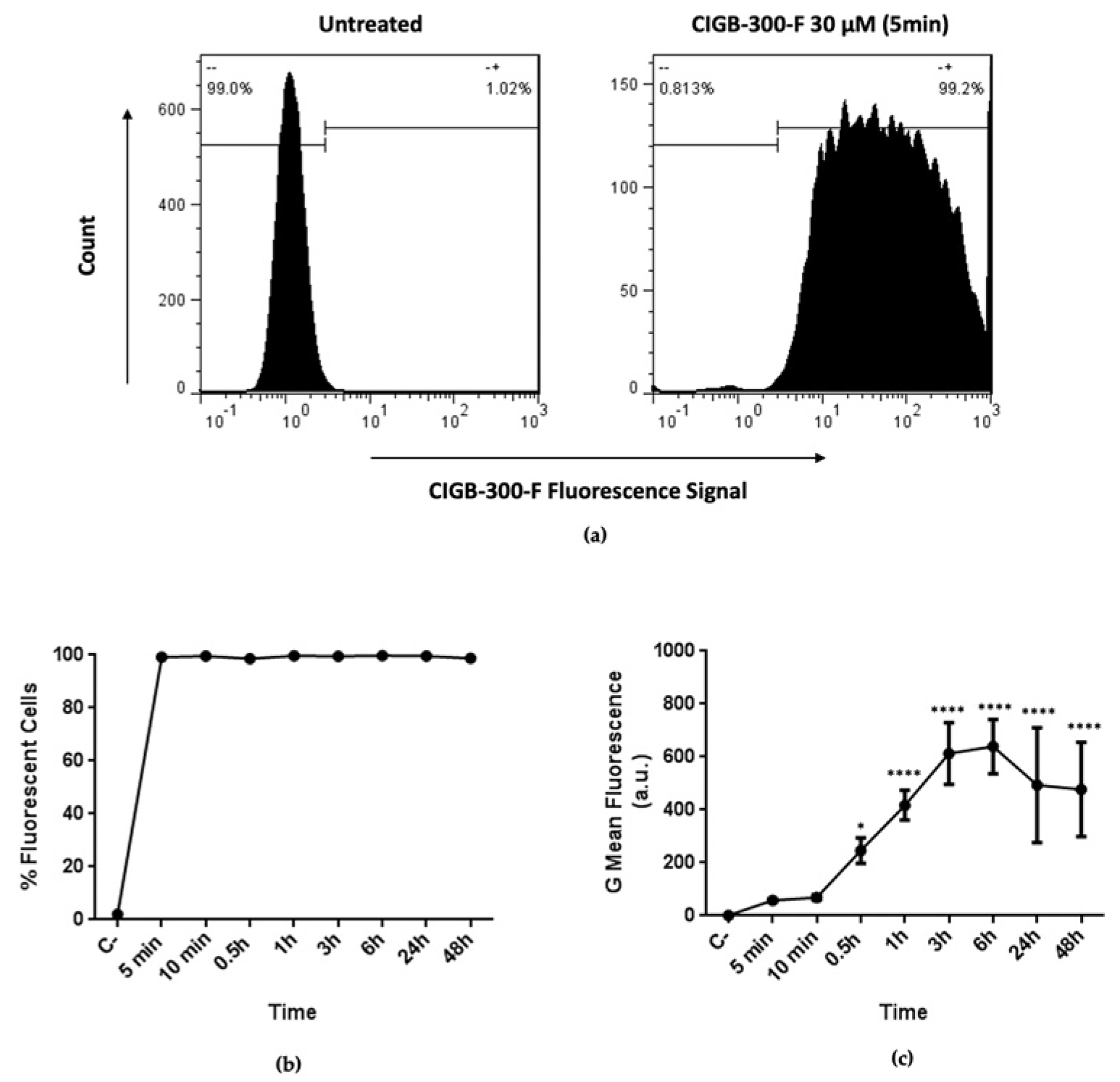
3.2. CIGB-300 Internalization and Interaction Profile in NCI-H460 Cells
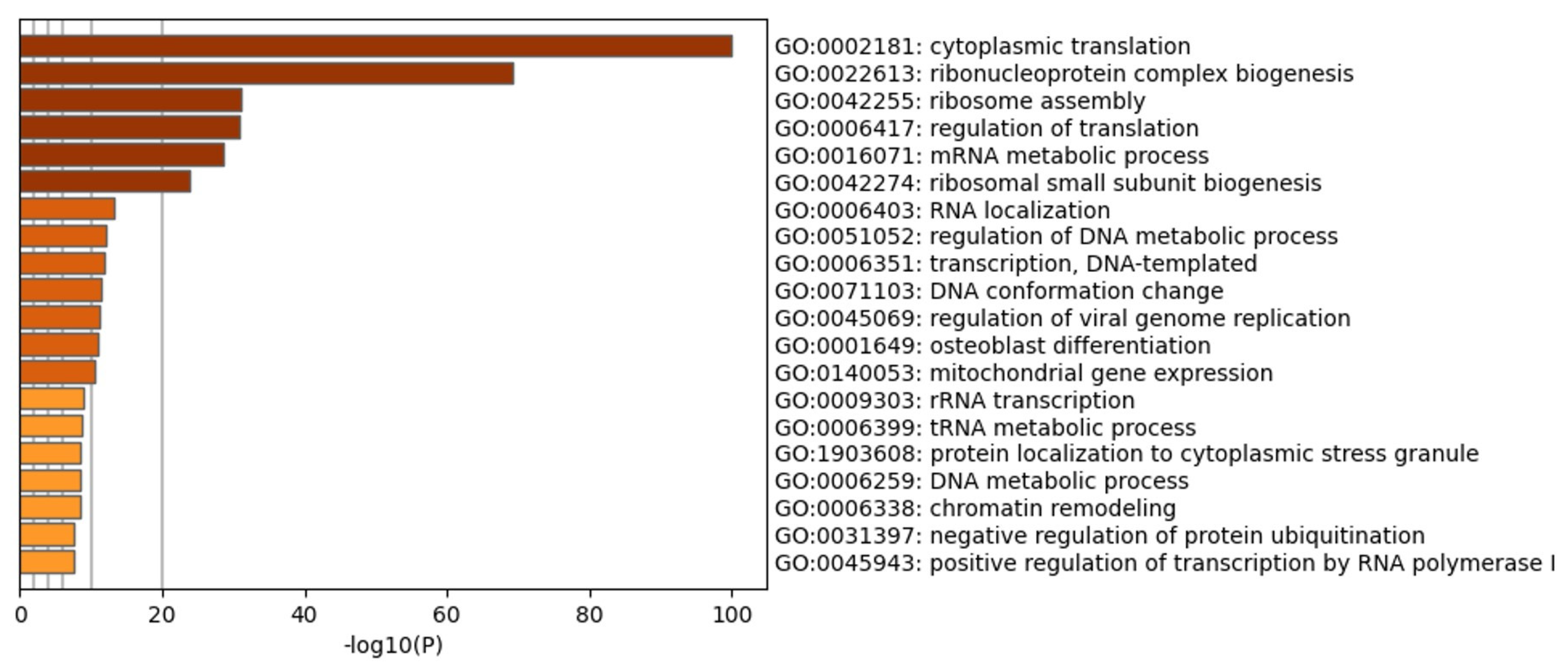
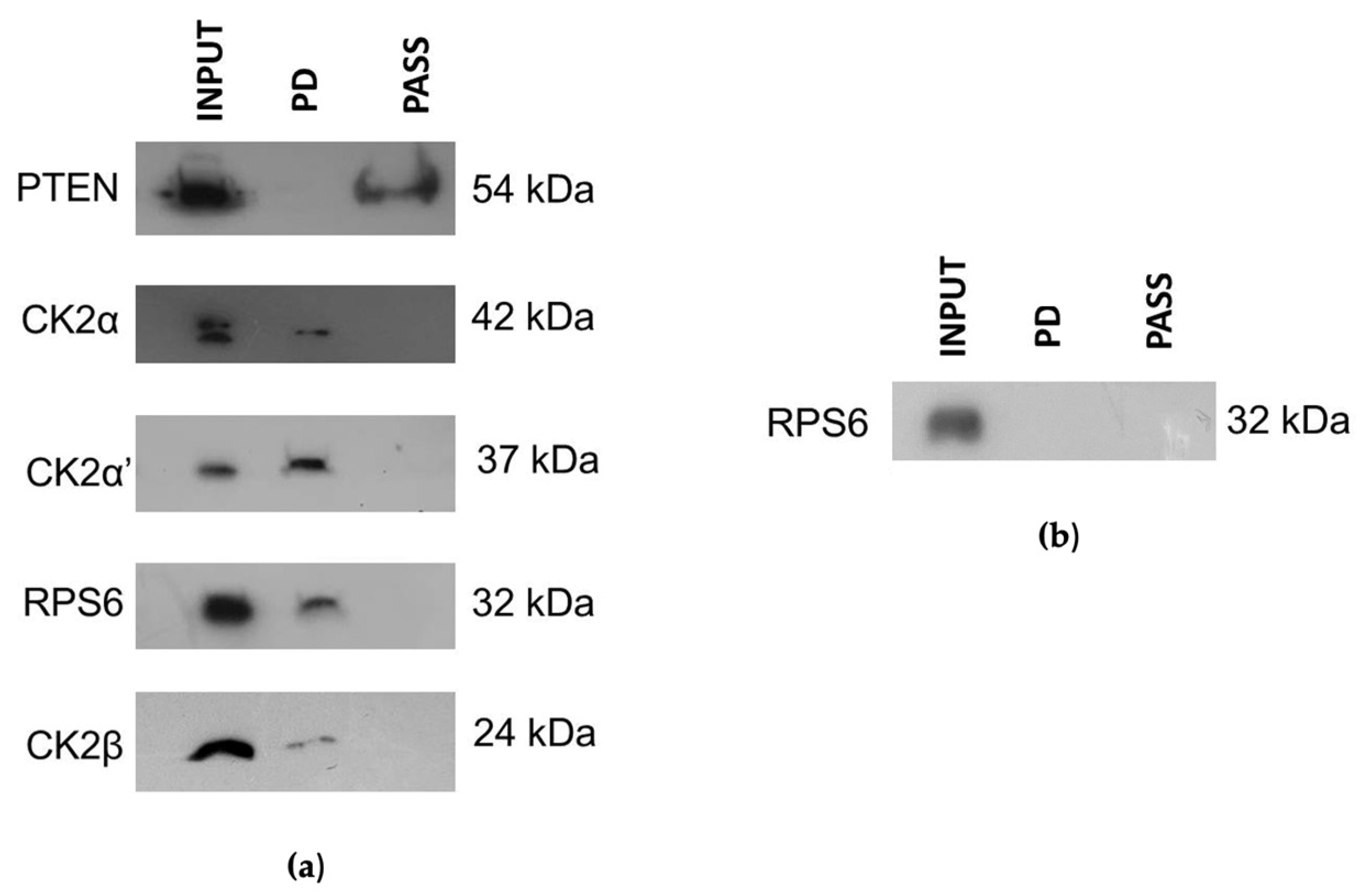
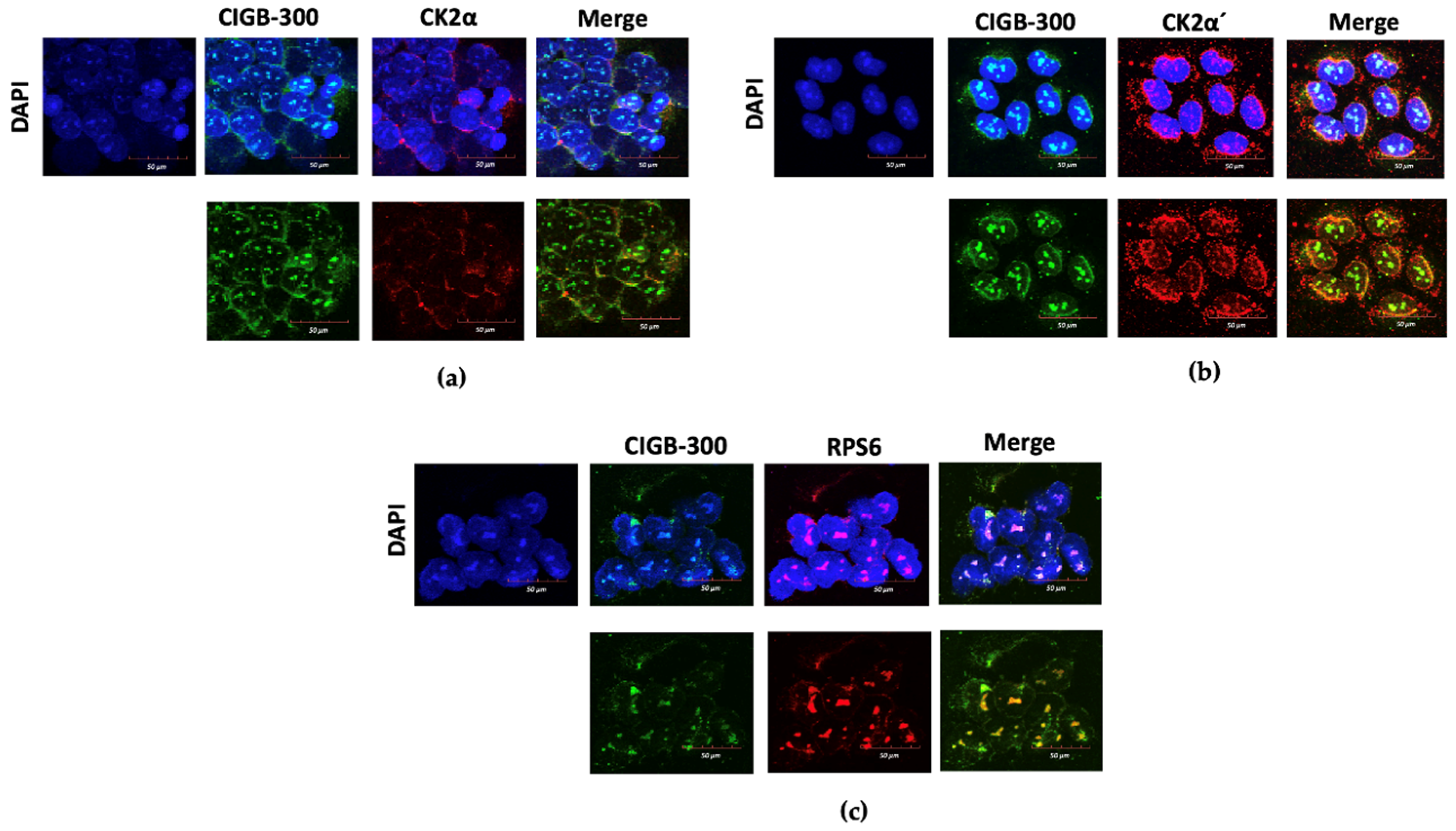
3.3. CIGB-300 Differentially Interacts with CK2 Subunits
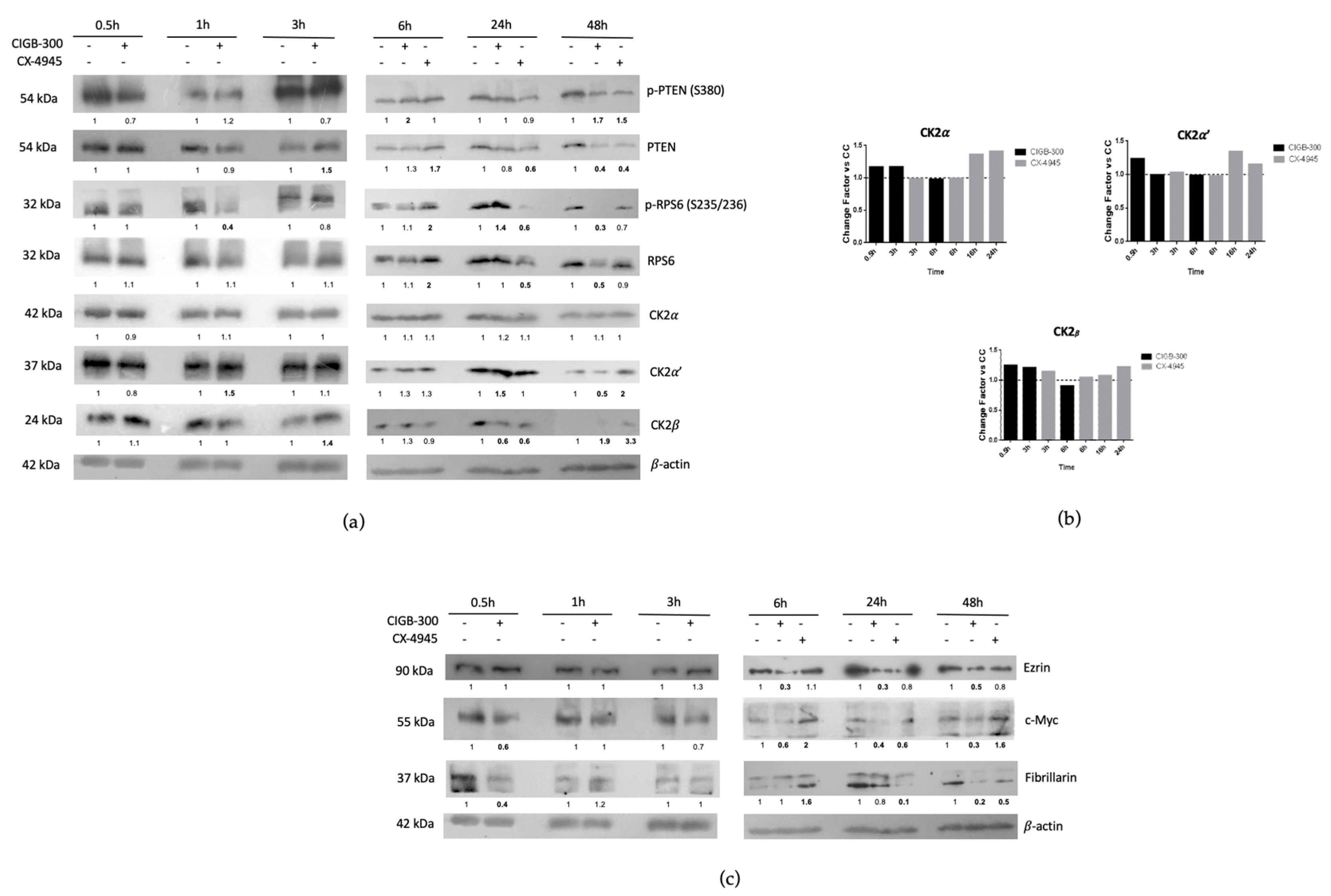
3.4. CIGB-300 Regulates Some CK2 Downstream Signaling Mediators in NCI-H460 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branco, H.; Oliveira, J.; Antunes, C.; Santos, L.L.; Vasconcelos, M.H.; Xavier, C.P.R. Pirfenidone Sensitizes NCI-H460 Non-Small Cell Lung Cancer Cells to Paclitaxel and to a Combination of Paclitaxel with Carboplatin. Int. J. Mol. Sci. 2022, 23, 3631. [Google Scholar] [CrossRef]
- Li, Q.; Li, K.; Yang, T.; Zhang, S.; Zhou, Y.; Li, Z.; Xiong, J.; Zhou, F.; Zhou, X.; Liu, L.; et al. Association of protein kinase CK2 inhibition with cellular radiosensitivity of non-small cell lung cancer. Sci. Rep. 2017, 7, 16134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.A.; Hughes, B.G. Targeted therapy for non-small cell lung cancer: Current standards and the promise of the future. Transl. Lung Cancer Res. 2015, 4, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Yao, J.; Li, B.; Shao, G.; Cui, Y. Inhibition of protein kinase CK2 sensitizes non-small cell lung cancer cells to cisplatin via upregulation of PML. Mol. Cell. Biochem. 2017, 436, 87–97. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef]
- Rajdev, K.; Siddiqui, A.H.; Ibrahim, U.; Patibandla, P.; Khan, T.; El-Sayegh, D. An Unusually Aggressive Large Cell Carcinoma of the Lung: Undiagnosed until Autopsy. Cureus 2018, 10, e2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Paradas, J.; Gómez-Sánchez, D.; Rosado, A.; Ucero, A.C.; Ferrer, I.; García-Luján, R.; Zugazagoitia, J.; Carrizo, N.; Enguita, A.B.; Conde, E.; et al. Comprehensive Characterization of Human Lung Large Cell Carcinoma Identifies Transcriptomic Signatures with Potential Implications in Response to Immunotherapy. J. Clin. Med. 2022, 11, 1500. [Google Scholar] [CrossRef]
- Jin, C.; Song, P.; Pang, J. The CK2 inhibitor CX4945 reverses cisplatin resistance in the A549/DDP human lung adenocarcinoma cell line. Oncol. Lett. 2019, 18, 3845–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, K.S.; Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Choi, C.M.; Chun, Y.J.; Lee, J.C. AKT/mTOR down-regulation by CX-4945, a CK2 inhibitor, promotes apoptosis in chemorefractory non-small cell lung cancer cells. Anticancer Res. 2015, 35, 1537–1542. [Google Scholar] [PubMed]
- Wang, Y.T.; Pan, S.H.; Tsai, C.F.; Kuo, T.C.; Hsu, Y.L.; Yen, H.Y.; Choong, W.K.; Wu, H.Y.; Liao, Y.C.; Hong, T.M.; et al. Phosphoproteomics Reveals HMGA1, a CK2 Substrate, as a Drug-Resistant Target in Non-Small Cell Lung Cancer. Sci. Rep. 2017, 7, 44021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Amin, E.B.; Mayo, M.W.; Chudgar, N.P.; Bucciarelli, P.R.; Kadota, K.; Adusumilli, P.S.; Jones, D.R. CK2α’ Drives Lung Cancer Metastasis by Targeting BRMS1 Nuclear Export and Degradation. Cancer Res. 2016, 76, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Chua, M.M.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Cirigliano, S.M.; Díaz Bessone, M.I.; Berardi, D.E.; Flumian, C.; Bal de Kier Joffé, E.D.; Perea, S.E.; Farina, H.G.; Todaro, L.B.; Urtreger, A.J. The synthetic peptide CIGB-300 modulates CK2-dependent signaling pathways affecting the survival and chemoresistance of non-small cell lung cancer cell lines. Cancer Cell Int. 2017, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Acero, F.B.; Capobianco, C.S.; Garona, J.; Cirigliano, S.M.; Perera, Y.; Urtreger, A.J.; Perea, S.E.; Alonso, D.F.; Farina, H.G. CIGB-300, an anti-CK2 peptide, inhibits angiogenesis, tumor cell invasion and metastasis in lung cancer models. Lung Cancer 2017, 107, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Uchino, J.; Goldmann, T.; Kimura, H. Editorial: Treatment for Non-Small Cell Lung Cancer in Distinct Patient Populations. Front. Oncol. 2022, 12, 838570. [Google Scholar] [CrossRef]
- Rivera-Concepcion, J.; Uprety, D.; Adjei, A.A. Challenges in the Use of Targeted Therapies in Non-Small Cell Lung Cancer. Cancer Res. Treat. 2022, 54, 315–329. [Google Scholar] [CrossRef]
- Zhang, S.; Long, H.; Yang, Y.L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2α down-regulates Notch1 signalling in lung cancer cells. J. Cell. Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Rosales, M.; Pérez, G.V.; Ramón, A.C.; Cruz, Y.; Rodríguez-Ulloa, A.; Besada, V.; Ramos, Y.; Vázquez-Blomquist, D.; Caballero, E.; Aguilar, D.; et al. Targeting of Protein Kinase CK2 in Acute Myeloid Leukemia Cells Using the Clinical-Grade Synthetic-Peptide CIGB-300. Biomedicines 2021, 9, 766. [Google Scholar] [CrossRef] [PubMed]
- Gober, M.K.; Flight, R.M.; Lambert, J.; Moseley, H.; Stromberg, A.; Black, E.P. Deregulation of a Network of mRNA and miRNA Genes Reveals That CK2 and MEK Inhibitors May Synergize to Induce Apoptosis KRAS-Active NSCLC. Cancer Inform. 2019, 18, 1176935119843507. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy—Potential clinical relevance. Cell. Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef]
- Imran, M.; Aslam Gondal, T.; Atif, M.; Shahbaz, M.; Batool Qaisarani, T.; Hanif Mughal, M.; Salehi, B.; Martorell, M.; Sharifi-Rad, J. Apigenin as an anticancer agent. Phytother. Res. 2020, 34, 1812–1828. [Google Scholar] [CrossRef]
- Meng, L.Q.; Liu, C.; Luo, Y.H.; Piao, X.J.; Wang, Y.; Zhang, Y.; Wang, J.R.; Wang, H.; Xu, W.T.; Liu, Y.; et al. Quinalizarin exerts an anti-tumour effect on lung cancer A549 cells by modulating the Akt, MAPK, STAT3 and p53 signalling pathways. Mol. Med. Rep. 2018, 17, 2626–2634. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Jia, H.; Li, L.; Zhang, G.; Zhao, M.; Cheng, Q.; Zheng, J.; Li, D. Inhibition of CK2 enhances UV-triggered apoptotic cell death in lung cancer cell lines. Oncol. Rep. 2013, 30, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Perera, Y.; Costales, H.C.; Diaz, Y.; Reyes, O.; Farina, H.G.; Mendez, L.; Gómez, R.E.; Acevedo, B.E.; Gomez, D.E.; Alonso, D.F.; et al. Sensitivity of tumor cells towards CIGB-300 anticancer peptide relies on its nucleolar localization. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2012, 18, 215–223. [Google Scholar] [CrossRef]
- Benavent Acero, F.R.; Perera Negrin, Y.; Alonso, D.F.; Perea, S.E.; Gomez, D.E.; Farina, H.G. Mechanisms of cellular uptake, intracellular transportation, and degradation of CIGB-300, a Tat-conjugated peptide, in tumor cell lines. Mol. Pharm. 2014, 11, 1798–1807. [Google Scholar] [CrossRef]
- Rosales, M.; Rodríguez-Ulloa, A.; Pérez, G.V.; Besada, V.; Soto, T.; Ramos, Y.; González, L.J.; Zettl, K.; Wiśniewski, J.R.; Yang, K.; et al. CIGB-300-Regulated Proteome Reveals Common and Tailored Response Patterns of AML Cells to CK2 Inhibition. Front. Mol. Biosci. 2022, 9, 834814. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales, M.; Rodríguez-Ulloa, A.; Besada, V.; Ramón, A.C.; Pérez, G.V.; Ramos, Y.; Guirola, O.; González, L.J.; Zettl, K.; Wiśniewski, J.R.; et al. Phosphoproteomic Landscape of AML Cells Treated with the ATP-Competitive CK2 Inhibitor CX-4945. Cells 2021, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Janin, M.; Coll-SanMartin, L.; Esteller, M. Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol. Cancer 2020, 19, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, Y.; Ramos, Y.; Padrón, G.; Caballero, E.; Guirola, O.; Caligiuri, L.G.; Lorenzo, N.; Gottardo, F.; Farina, H.G.; Filhol, O.; et al. CIGB-300 anticancer peptide regulates the protein kinase CK2-dependent phosphoproteome. Mol. Cell. Biochem. 2020, 470, 63–75. [Google Scholar] [CrossRef]
- Perera, Y.; Melão, A.; Ramón, A.C.; Vázquez, D.; Ribeiro, D.; Perea, S.E.; Barata, J.T. Clinical-Grade Peptide-Based Inhibition of CK2 Blocks Viability and Proliferation of T-ALL Cells and Counteracts IL-7 Stimulation and Stromal Support. Cancers 2020, 12, 1377. [Google Scholar] [CrossRef]
- Yi, Y.W.; You, K.S.; Park, J.S.; Lee, S.G.; Seong, Y.S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2021, 23, 48. [Google Scholar] [CrossRef]
- Alcaraz, E.; Vilardell, J.; Borgo, C.; Sarró, E.; Plana, M.; Marin, O.; Pinna, L.A.; Bayascas, J.R.; Meseguer, A.; Salvi, M.; et al. Effects of CK2β subunit down-regulation on Akt signalling in HK-2 renal cells. PloS ONE 2020, 15, e0227340. [Google Scholar] [CrossRef] [Green Version]
- Husain, K.; Williamson, T.T.; Nelson, N.; Ghansah, T. Protein kinase 2 (CK2): A potential regulator of immune cell development and function in cancer. Immunol. Med. 2021, 44, 159–174. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, H.P.; Duan, H.F.; Gao, L.H.; Shao, Y.; Chen, K.Y.; Wang, Y.L.; Lan, F.H.; Hu, X.W. Aggregation of Ribosomal Protein S6 at Nucleolus Is Cell Cycle-Controlled and Its Function in Pre-rRNA Processing Is Phosphorylation Dependent. J. Cell. Biochem. 2016, 117, 1649–1657. [Google Scholar] [CrossRef]
- Li, J.; Wei, K.; Yu, H.; Jin, D.; Wang, G.; Yu, B. Prognostic Value of Ezrin in Various Cancers: A Systematic Review and Updated Meta-analysis. Sci. Rep. 2015, 5, 17903. [Google Scholar] [CrossRef]
- Lee, H.W.; Kim, E.H.; Oh, M.H. Clinicopathologic implication of ezrin expression in non-small cell lung cancer. Korean J. Pathol. 2012, 46, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Yaylim, I.; Ozkan, N.E.; Isitmangil, T.; Isitmangil, G.; Turna, A.; Isbir, T. CK2 enzyme affinity against c-myc 424-434 substrate in human lung cancer tissue. Asian Pac. J. Cancer Prev. APJCP 2012, 13, 5233–5236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, A.N.; Yang, J.M.; Kim, H.; Jheon, S.; Kim, K.; Lee, C.T.; Jin, Y.; Yun, S.; Chung, J.H.; Paik, J.H. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. Br. J. Cancer 2014, 110, 2688–2699. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Rapp, U.R.; Korn, C.; Ceteci, F.; Karreman, C.; Luetkenhaus, K.; Serafin, V.; Zanucco, E.; Castro, I.; Potapenko, T. MYC is a metastasis gene for non-small-cell lung cancer. PloS ONE 2009, 4, e6029. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Yang, J.; Yi, J. Nucleolar Stress: Hallmarks, sensing mechanism and diseases. Cell Stress 2018, 2, 125–140. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, G.; Li, Q.; Xie, F. Increased fibrillarin expression is associated with tumor progression and an unfavorable prognosis in hepatocellular carcinoma. Oncol. Lett. 2021, 21, 92. [Google Scholar] [CrossRef]
- Shubina, M.Y.; Musinova, Y.R.; Sheval, E.V. Proliferation, cancer, and aging-novel functions of the nucleolar methyltransferase fibrillarin? Cell Biol. Int. 2018, 42, 1463–1466. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot ACC | Gene Names | Protein Names |
|---|---|---|
| Q8NE71 | ABCF1 | ATP-binding cassette sub-family F member 1 |
| P20290 | BTF3 | Transcription factor BTF3 |
| Q07021 | C1QBP | Complement component 1 Q subcomponent-binding protein, mitochondrial |
| P68400 | CSNK2A1 | Casein kinase II subunit alpha |
| P35659 | DEK | Protein DEK |
| P20042 | EIF2S2 | Eukaryotic translation initiation factor 2 subunit 2 |
| Q13283 | G3BP1 | Ras GTPase-activating protein-binding protein 1 |
| Q92769 | HDAC2 | Histone deacetylase 2 |
| P62805 | HIST1H4A | Histone H4 |
| P09651 | HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 processed |
| P22626 | HNRNPA2B1 | Heterogeneous nuclear ribonucleoproteins A2/B1 |
| P07910 | HNRNPC | Heterogeneous nuclear ribonucleoproteins C1/C2 |
| Q16666 | IFI16 | Gamma-interferon-inducible protein 16 |
| P46821 | MAP1B | Microtubule-associated protein 1B |
| Q9UKD2 | MRTO4 | mRNA turnover protein 4 homolog |
| P19338 | NCL | Nucleolin |
| Q14978 | NOLC1 | Nucleolar and coiled-body phosphoprotein 1 |
| P06748 | NPM1 | Nucleophosmin |
| Q13393 | PLD1 | Phospholipase D1 |
| Q15287 | RNPS1 | RNA-binding protein with serine-rich domain 1 |
| P46777 | RPL5 | 60S ribosomal protein L5 |
| Q96SB4 | SRPK1 | SRSF protein kinase 1 |
| P05455 | SSB | Lupus La protein |
| P43307 | SSR1 | Translocon-associated protein subunit alpha |
| Q08945 | SSRP1 | FACT complex subunit SSRP1 |
| P53999 | SUB1 | Activated RNA polymerase II transcriptional coactivator p15 |
| Q13428 | TCOF1 | Treacle protein |
| P11387 | TOP1 | DNA topoisomerase 1 |
| P11388 | TOP2A | DNA topoisomerase 2-alpha |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez, G.V.; Rosales, M.; Ramón, A.C.; Rodríguez-Ulloa, A.; Besada, V.; González, L.J.; Aguilar, D.; Vázquez-Blomquist, D.; Falcón, V.; Caballero, E.; et al. CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model. Biomedicines 2023, 11, 43. https://doi.org/10.3390/biomedicines11010043
Pérez GV, Rosales M, Ramón AC, Rodríguez-Ulloa A, Besada V, González LJ, Aguilar D, Vázquez-Blomquist D, Falcón V, Caballero E, et al. CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model. Biomedicines. 2023; 11(1):43. https://doi.org/10.3390/biomedicines11010043
Chicago/Turabian StylePérez, George V., Mauro Rosales, Ailyn C. Ramón, Arielis Rodríguez-Ulloa, Vladimir Besada, Luis J. González, Daylen Aguilar, Dania Vázquez-Blomquist, Viviana Falcón, Evelin Caballero, and et al. 2023. "CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model" Biomedicines 11, no. 1: 43. https://doi.org/10.3390/biomedicines11010043
APA StylePérez, G. V., Rosales, M., Ramón, A. C., Rodríguez-Ulloa, A., Besada, V., González, L. J., Aguilar, D., Vázquez-Blomquist, D., Falcón, V., Caballero, E., Carvalho, P. C., Caldeira, R. S., Yang, K., Perera, Y., & Perea, S. E. (2023). CIGB-300 Anticancer Peptide Differentially Interacts with CK2 Subunits and Regulates Specific Signaling Mediators in a Highly Sensitive Large Cell Lung Carcinoma Cell Model. Biomedicines, 11(1), 43. https://doi.org/10.3390/biomedicines11010043