
Enhancing Muscle Intracellular Ca2+ Homeostasis and Glucose Uptake: Passive Pulsatile Shear Stress Treatment in Type 2 Diabetes



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. pGz Protocol
2.3. Double-Barreled Ca2+-Selective Microelectrodes
2.4. Recording of [Ca2+]i and Glucose Uptake in Muscle Fibers In Vivo
2.5. Determination of ROS in Muscle Cells
2.6. Creatine Kinase Serum Level Determination
2.7. Measurement of Plasma Interleukin and TNF-α Concentrations
2.8. Muscle Functional Testings
2.9. N-nitro-L-arginine Methyl Ester (L-NAME) Protocol
2.10. Glucose Determinations
2.11. Western Blotting and Protein Expression
2.12. Solutions
2.13. Statistical Analysis
3. Results
3.1. pGz Reduced [Ca2+]i and Increase Glucose Uptake in db/db Skeletal Muscle
3.2. Effect of pGz on ROS Production in db/db Muscle Fibers
3.3. Reduction in Muscle Injury by pGz in db/db Mice
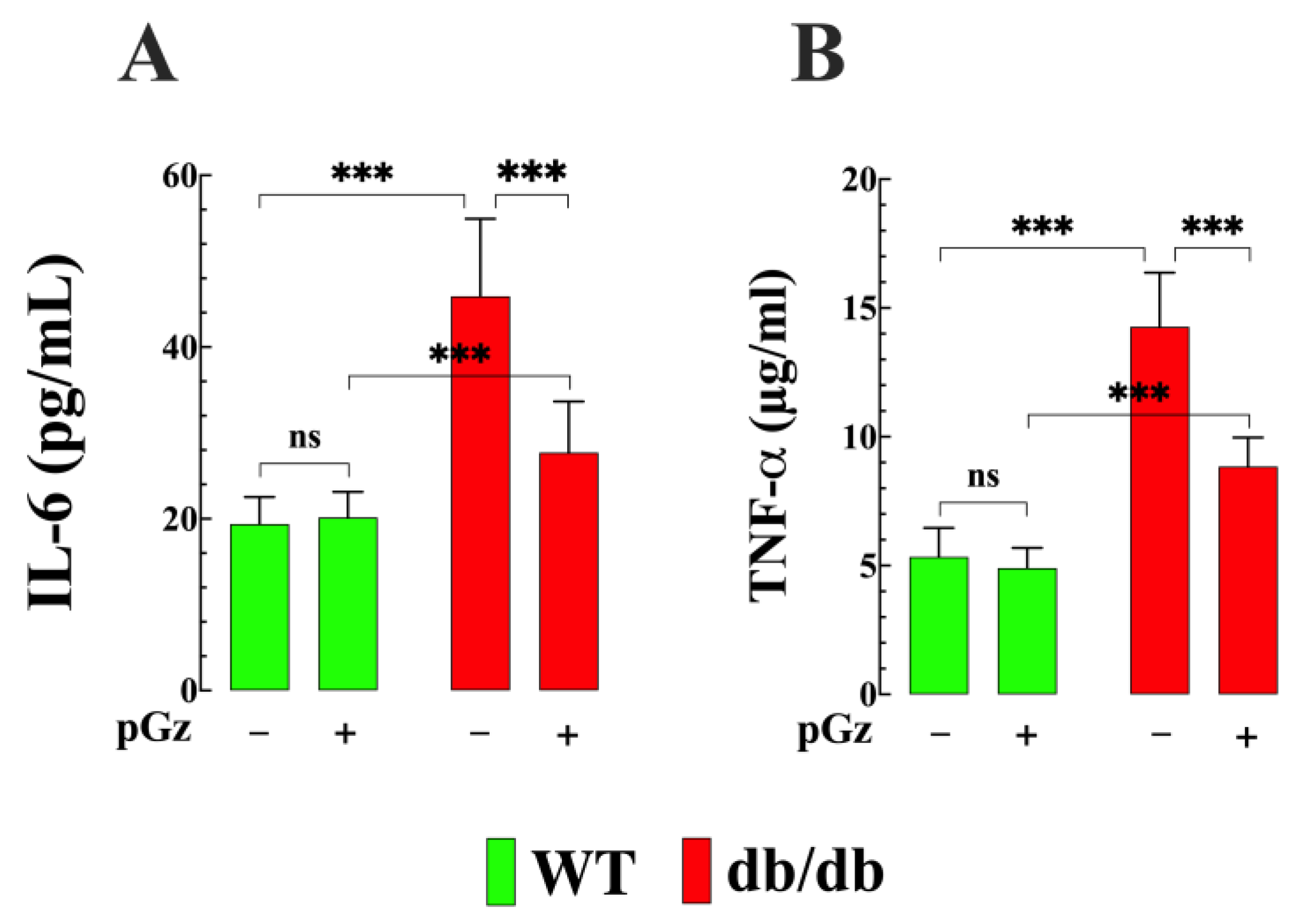
3.4. Effects of pGz on Plasma Interleukin 6 and TNF-α Concentrations in db/db Mice
3.5. pGz Improve Muscle Funtion in Diabetic Mice
3.6. The Role of NO in the Cytoprotective Effects of pGz
3.7. Effect of pGz on Blood Glucose Level
3.8. Effect of pGz on the Expression of the NOS Protein in db/db Muscle
4. Discussion
- The application of pGz effectively reduced the [Ca2+]i overload and increased the glucose uptake observed in db/db skeletal muscle fibers;
- The study revealed that db/db muscle exhibited elevated intracellular ROS production, and treatment with pGz effectively reduced this elevated ROS production;
- The serum levels of CK in db/db mice were significantly higher compared to WT mice, and pGz intervention reduced CK levels in db/db mice;
- There was a significant increase in pro-inflammatory cytokines, specifically IL-6 TNF-α, in db/db mice; pGz treatment decreased the plasma concentrations of both IL-6 and TNF-α in db/db mice;
- db/db muscles showed greater fatigability and reduced force compared to WT; pGz treatment resulted in a reduction of muscle fatigability and improvement of muscle force;
- L-NAME pretreatment abolished the cytoprotective effect of pGz on [Ca2+]I and ROS production in db/db mice;
- The expression of eNOS and p-eNOS proteins in db/db muscle was reduced, while iNOS was enhanced. Treatment with pGz reversed the expression pattern observed in db/db mice by increasing eNOS, p-eNOS, and nNOS expression while causing a decrease in iNOS levels in db/db skeletal muscle.
4.1. pGz Reduces Intracellular Ca2+ and Improves Glucose Uptake
4.2. Intracellular ROS Is Reduced by pGz in db/db Mice
4.3. Reduction of Muscle Damage by pGz in db/db Mice
4.4. Decrease Inflammatory Cytokines by pGz
4.5. pGz Reduces Muscle Fatigability and Increases Strength in db/db Mice
4.6. Effects of pGz on NOS Expression in db/db Muscles
4.7. Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Ramtahal, R.; Khan, C.; Maharaj-Khan, K.; Nallamothu, S.; Hinds, A.; Dhanoo, A.; Yeh, H.C.; Hill-Briggs, F.; Lazo, M. Prevalence of self-reported sleep duration and sleep habits in type 2 diabetes patients in South Trinidad. J. Epidemiol. Glob. Health 2015, 5, S35–S43. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Solomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef]
- Uryash, A.; Mijares, A.; Lopez, C.E.; Adams, J.A.; Lopez, J.R. Chronic Elevation of Skeletal Muscle [Ca2+]i Impairs Glucose Uptake. An in Vivo and in Vitro Study. Front. Physiol. 2022, 13, 872624. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Ehrlich, B.E. Signaling in muscle contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef]
- Cali, T.; Brini, M.; Carafoli, E. Regulation of Cell Calcium and Role of Plasma Membrane Calcium ATPases. Int. Rev. Cell Mol. Biol. 2017, 332, 259–296. [Google Scholar] [CrossRef]
- Uryash, A.; Bassuk, J.; Kurlansky, P.; Altamirano, F.; Lopez, J.R.; Adams, J.A. Antioxidant Properties of Whole Body Periodic Acceleration (pGz). PLoS ONE 2015, 10, e0131392. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef]
- Uryash, A.; Mijares, A.; Esteve, E.; Adams, J.A.; Lopez, J.R. Cardioprotective Effect of Whole Body Periodic Acceleration in Dystrophic Phenotype mdx Rodent. Front. Physiol. 2021, 12, 658042. [Google Scholar] [CrossRef]
- Eltit, J.M.; Ding, X.; Pessah, I.N.; Allen, P.D.; Lopez, J.R. Nonspecific sarcolemmal cation channels are critical for the pathogenesis of malignant hyperthermia. FASEB J. 2013, 27, 991–1000. [Google Scholar] [CrossRef]
- Furfine, E.S.; Carbine, K.; Bunker, S.; Tanoury, G.; Harmon, M.; Laubach, V.; Sherman, P. Potent inhibition of human neuronal nitric oxide synthase by N(G)-nitro-L-arginine methyl ester results from contaminating N(G)-nitro-L-arginine. Life Sci. 1997, 60, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Wang, Y.; Shen, Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J. Biochem. Biophys. Methods 2005, 64, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Fuentealba, C.; Contreras-Ferrat, A.E.; Altamirano, F.; Espinosa, A.; Li, Q.; Niu, W.; Lavandero, S.; Klip, A.; Jaimovich, E. Electrical stimuli release ATP to increase GLUT4 translocation and glucose uptake via PI3Kgamma-Akt-AS160 in skeletal muscle cells. Diabetes 2013, 62, 1519–1526. [Google Scholar] [CrossRef]
- Darenskaya, M.A.; Kolesnikova, L.I.; Kolesnikov, S.I. Oxidative Stress: Pathogenetic Role in Diabetes Mellitus and Its Complications and Therapeutic Approaches to Correction. Bull. Exp. Biol. Med. 2021, 171, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Hassan-Zadeh, V. IL-6 signalling pathways and the development of type 2 diabetes. Inflammopharmacology 2018, 26, 685–698. [Google Scholar] [CrossRef]
- Yang, J.; Park, Y.; Zhang, H.; Xu, X.; Laine, G.A.; Dellsperger, K.C.; Zhang, C. Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1850–H1858. [Google Scholar] [CrossRef]
- Nesti, L.; Pugliese, N.R.; Sciuto, P.; Natali, A. Type 2 diabetes and reduced exercise tolerance: A review of the literature through an integrated physiology approach. Cardiovasc. Diabetol. 2020, 19, 134. [Google Scholar] [CrossRef]
- Orlando, G.; Sacchetti, M.; D’Errico, V.; Haxhi, J.; Rapisarda, G.; Pugliese, G.; Balducci, S. Muscle fatigability in patients with type 2 diabetes: Relation with long-term complications. Diabetes Metab. Res. Rev. 2020, 36, e3231. [Google Scholar] [CrossRef]
- Ruze, R.; Liu, T.; Zou, X.; Song, J.; Chen, Y.; Xu, R.; Yin, X.; Xu, Q. Obesity and type 2 diabetes mellitus: Connections in epidemiology, pathogenesis, and treatments. Front. Endocrinol. 2023, 14, 1161521. [Google Scholar] [CrossRef]
- Clodi, M.; Toplak, H.; Resl, M.; Brix, J.; Leitner, D.R.; Harreiter, J.; Hoppichler, F.; Wascher, T.C.; Schindler, K.; Ludvik, B. Obesity and type 2 diabetes (Update 2023). Wien. Klin. Wochenschr. 2023, 135, 91–97. [Google Scholar] [CrossRef]
- Harreiter, J.; Roden, M. Diabetes mellitus: Definition, classification, diagnosis, screening and prevention (Update 2023). Wien. Klin. Wochenschr. 2023, 135, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Tinajero, M.G.; Malik, V.S. An Update on the Epidemiology of Type 2 Diabetes: A Global Perspective. Endocrinol. Metab. Clin. N. Am. 2021, 50, 337–355. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, S.Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z. Muscle Insulin Resistance and the Inflamed Microvasculature: Fire from Within. Int. J. Mol. Sci. 2019, 20, 562. [Google Scholar] [CrossRef]
- De Mario, A.; Gherardi, G.; Rizzuto, R.; Mammucari, C. Skeletal muscle mitochondria in health and disease. Cell Calcium 2021, 94, 102357. [Google Scholar] [CrossRef]
- Creismeas, A.; Gazaille, C.; Bourdon, A.; Lallemand, M.A.; Francois, V.; Allais, M.; Ledevin, M.; Larcher, T.; Toumaniantz, G.; Lafoux, A.; et al. TRPC3, but not TRPC1, as a good therapeutic target for standalone or complementary treatment of DMD. J. Transl. Med. 2021, 19, 519. [Google Scholar] [CrossRef]
- Meszaros, L.G.; Minarovic, I.; Zahradnikova, A. Inhibition of the skeletal muscle ryanodine receptor calcium release channel by nitric oxide. FEBS Lett. 1996, 380, 49–52. [Google Scholar] [CrossRef]
- Meszaros, L.G. Nitric oxide and the ryanodine receptor Ca-release channel. Circ. Res. 2010, 107, e1. [Google Scholar] [CrossRef] [PubMed]
- Snider, B.J.; Choi, J.; Turetsky, D.M.; Canzoniero, L.M.; Sensi, S.L.; Sheline, C.T.; Wang, X.; Yu, S.P.; Choi, D.W. Nitric oxide reduces Ca2+ and Zn2+ influx through voltage-gated Ca2+ channels and reduces Zn2+ neurotoxicity. Neuroscience 2000, 100, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sul, J.Y.; Haydon, P.G. A calcium-induced calcium influx factor, nitric oxide, modulates the refilling of calcium stores in astrocytes. J. Neurosci. 2003, 23, 10302–10310. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, W.; Kazama, S.; Suzuki, T.; Yamana, R.; Miyazaki, Y.; Tanaka, K.; Usami, M.; Takizawa, T. L-NAME, a nitric oxide synthase inhibitor, increases the protein expression of both executioner and inhibitor of apoptosis in the placental bed of mid-to-late pregnant rats. Congenit. Anom. 2021, 61, 183–187. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Leopold, E.; Schmidt, K.; Brunner, F.; Mayer, B. Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): Requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br. J. Pharmacol. 1996, 118, 1433–1440. [Google Scholar] [CrossRef]
- Draznin, B.; Lewis, D.; Houlder, N.; Sherman, N.; Adamo, M.; Garvey, W.T.; LeRoith, D.; Sussman, K. Mechanism of insulin resistance induced by sustained levels of cytosolic free calcium in rat adipocytes. Endocrinology 1989, 125, 2341–2349. [Google Scholar] [CrossRef]
- Draznin, B.; Sussman, K.E.; Eckel, R.H.; Kao, M.; Yost, T.; Sherman, N.A. Possible role of cytosolic free calcium concentrations in mediating insulin resistance of obesity and hyperinsulinemia. J. Clin. Investig. 1988, 82, 1848–1852. [Google Scholar] [CrossRef]
- Nishiki, Y.; Kono, T.; Fukao, K.; Sato, K.; Takahashi, K.; Toyomizu, M.; Akiba, Y. Nitric oxide (NO) is involved in modulation of non-insulin mediated glucose transport in chicken skeletal muscles. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 149, 101–107. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, X.; Zong, B.; Yuan, H.; Wang, Z.; Wei, Y.; Wang, X.; Liu, G.; Zhang, J.; Li, S.; et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J. Cell Mol. Med. 2018, 22, 4437–4448. [Google Scholar] [CrossRef]
- Huang, X.; Sun, M.; Li, D.; Liu, J.; Guo, H.; Dong, Y.; Jiang, L.; Pan, Q.; Man, Y.; Wang, S.; et al. Augmented NADPH oxidase activity and p22phox expression in monocytes underlie oxidative stress of patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2011, 91, 371–380. [Google Scholar] [CrossRef]
- Dyakova, E.Y.; Kapilevich, L.V.; Shylko, V.G.; Popov, S.V.; Anfinogenova, Y. Physical exercise associated with NO production: Signaling pathways and significance in health and disease. Front. Cell Dev. Biol. 2015, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Kohnert, K.D.; Freyse, E.J.; Salzsieder, E. Glycaemic variability and pancreatic beta-cell dysfunction. Curr. Diabetes Rev. 2012, 8, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria and ageing: Role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369. [Google Scholar] [CrossRef]
- Frank, M.; Finsterer, J. Creatine kinase elevation, lactacidemia, and metabolic myopathy in adult patients with diabetes mellitus. Endocr. Pract. 2012, 18, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Jevric-Causevic, A.; Malenica, M.; Dujic, T. Creatine kinase activity in patients with diabetes mellitus type I and type II. Bosn. J. Basic. Med. Sci. 2006, 6, 5–9. [Google Scholar] [CrossRef]
- Hansen, D.; Dendale, P.; Beelen, M.; Jonkers, R.A.; Mullens, A.; Corluy, L.; Meeusen, R.; van Loon, L.J. Plasma adipokine and inflammatory marker concentrations are altered in obese, as opposed to non-obese, type 2 diabetes patients. Eur. J. Appl. Physiol. 2010, 109, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Hossain, M.; Mathews, C.; Martinez, P.; Pino, P.; Gay, J.L.; Rentfro, A.; McCormick, J.B.; Fisher-Hoch, S.P. Type 2-diabetes is associated with elevated levels of TNF-alpha, IL-6 and adiponectin and low levels of leptin in a population of Mexican Americans: A cross-sectional study. Cytokine 2012, 57, 136–142. [Google Scholar] [CrossRef]
- Saxena, M.; Srivastava, N.; Banerjee, M. Association of IL-6, TNF-alpha and IL-10 gene polymorphisms with type 2 diabetes mellitus. Mol. Biol. Rep. 2013, 40, 6271–6279. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Liu, C.; Feng, X.; Li, Q.; Wang, Y.; Li, Q.; Hua, M. Adiponectin, TNF-alpha and inflammatory cytokines and risk of type 2 diabetes: A systematic review and meta-analysis. Cytokine 2016, 86, 100–109. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Zhang, H.; Hill, M.A.; Zhang, C.; Park, Y. Interaction of IL-6 and TNF-alpha contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS ONE 2017, 12, e0187189. [Google Scholar] [CrossRef]
- Adams, J.A.; Lopez, J.R.; Uryash, A.; Sackner, M.A. Whole body periodic acceleration (pGz) improves endotoxin induced cardiomyocyte contractile dysfunction and attenuates the inflammatory response in mice. Heliyon 2021, 7, e06444. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Bruunsgaard, H.; Weis, N.; Hendel, H.W.; Andreassen, B.U.; Eldrup, E.; Dela, F.; Pedersen, B.K. Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech. Ageing Dev. 2003, 124, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Goodpaster, B.H.; Lee, J.S.; Kuller, L.H.; Boudreau, R.; de Rekeneire, N.; Harris, T.B.; Kritchevsky, S.; Tylavsky, F.A.; Nevitt, M.; et al. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 2009, 32, 1993–1997. [Google Scholar] [CrossRef] [PubMed]
- Leenders, M.; Verdijk, L.B.; van der Hoeven, L.; Adam, J.J.; van Kranenburg, J.; Nilwik, R.; van Loon, L.J. Patients with type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J. Am. Med. Dir. Assoc. 2013, 14, 585–592. [Google Scholar] [CrossRef]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Alvares, T.S.; Conte, C.A.; Paschoalin, V.M.; Silva, J.T.; Meirelles Cde, M.; Bhambhani, Y.N.; Gomes, P.S. Acute l-arginine supplementation increases muscle blood volume but not strength performance. Appl. Physiol. Nutr. Metab. 2012, 37, 115–126. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Roy, D.; Perreault, M.; Marette, A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am. J. Physiol. 1998, 274, E692–E699. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Ghasemi, A. Role of Nitric Oxide in Insulin Secretion and Glucose Metabolism. Trends Endocrinol. Metab. 2020, 31, 118–130. [Google Scholar] [CrossRef]
- Gheibi, S.; Ghasemi, A. Insulin secretion: The nitric oxide controversy. EXCLI J. 2020, 19, 1227–1245. [Google Scholar] [CrossRef]
- Shankar, R.R.; Wu, Y.; Shen, H.Q.; Zhu, J.S.; Baron, A.D. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes 2000, 49, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Soskic, S.S.; Dobutovic, B.D.; Sudar, E.M.; Obradovic, M.M.; Nikolic, D.M.; Djordjevic, J.D.; Radak, D.J.; Mikhailidis, D.P.; Isenovic, E.R. Regulation of Inducible Nitric Oxide Synthase (iNOS) and its Potential Role in Insulin Resistance, Diabetes and Heart Failure. Open Cardiovasc. Med. J. 2011, 5, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Sugita, H.; Fujimoto, M.; Yasukawa, T.; Shimizu, N.; Sugita, M.; Yasuhara, S.; Martyn, J.A.; Kaneki, M. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J. Biol. Chem. 2005, 280, 14203–14211. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.H.; De Sanctis, J.B.; de Briceno, L.M.; Hernandez, N.; Finol, H.J. Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J. Endocrinol. 2004, 181, 419–427. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H.; Kang, G. Nitrosative Stress and Human Disease: Therapeutic Potential of Denitrosylation. Int. J. Mol. Sci. 2021, 22, 9794. [Google Scholar] [CrossRef]
- Carvalho-Filho, M.A.; Ueno, M.; Hirabara, S.M.; Seabra, A.B.; Carvalheira, J.B.; de Oliveira, M.G.; Velloso, L.A.; Curi, R.; Saad, M.J. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: A novel mechanism of insulin resistance. Diabetes 2005, 54, 959–967. [Google Scholar] [CrossRef]
- Kaneki, M.; Shimizu, N.; Yamada, D.; Chang, K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox Signal 2007, 9, 319–329. [Google Scholar] [CrossRef]
- Suzuki, H.; Colasanti, M. Cross-talk between constitutive and inducible nitric oxide synthases. Circulation 2001, 103, E81. [Google Scholar] [CrossRef]
- Tessari, P.; Cecchet, D.; Cosma, A.; Vettore, M.; Coracina, A.; Millioni, R.; Iori, E.; Puricelli, L.; Avogaro, A.; Vedovato, M. Nitric oxide synthesis is reduced in subjects with type 2 diabetes and nephropathy. Diabetes 2010, 59, 2152–2159. [Google Scholar] [CrossRef]
- Kumar, D.; Shankar, K.; Patel, S.; Gupta, A.; Varshney, S.; Gupta, S.; Rajan, S.; Srivastava, A.; Vishwakarma, A.L.; Gaikwad, A.N. Chronic hyperinsulinemia promotes meta-inflammation and extracellular matrix deposition in adipose tissue: Implications of nitric oxide. Mol. Cell Endocrinol. 2018, 477, 15–28. [Google Scholar] [CrossRef]
- Horton, R.A.; Ceppi, E.D.; Knowles, R.G.; Titheradge, M.A. Inhibition of hepatic gluconeogenesis by nitric oxide: A comparison with endotoxic shock. Biochem. J. 1994, 299, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Zhu, J.S.; Marshall, S.; Irsula, O.; Brechtel, G.; Keech, C. Insulin resistance after hypertension induced by the nitric oxide synthesis inhibitor L-NMMA in rats. Am. J. Physiol. 1995, 269, E709–E715. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A.; Mangino, M.J.; Bassuk, J.; Kurlansky, P.; Sackner, M.A. Regional blood flow during periodic acceleration. Crit. Care Med. 2001, 29, 1983–1988. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Abraham, W.M.; Ahmed, A.; Serebriakov, I.; Lauredo, I.T.; Bassuk, J.; Adams, J.A.; Sackner, M.A. Whole-body periodic acceleration modifies experimental asthma in sheep. Am. J. Respir. Crit. Care Med. 2006, 174, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.A.; Uryash, A.; Lopez, J.R.; Sackner, M.A. Whole body periodic acceleration improves survival and microvascular leak in a murine endotoxin model. PLoS ONE 2019, 14, e0208681. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.L.; Johansson, S.; Wallander, M.A.; Rodriguez, L.A. Trends in the prevalence and incidence of diabetes in the UK: 1996–2005. J. Epidemiol. Community Health 2009, 63, 332–336. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uryash, A.; Umlas, J.; Mijares, A.; Adams, J.A.; Lopez, J.R. Enhancing Muscle Intracellular Ca2+ Homeostasis and Glucose Uptake: Passive Pulsatile Shear Stress Treatment in Type 2 Diabetes. Biomedicines 2023, 11, 2596. https://doi.org/10.3390/biomedicines11102596
Uryash A, Umlas J, Mijares A, Adams JA, Lopez JR. Enhancing Muscle Intracellular Ca2+ Homeostasis and Glucose Uptake: Passive Pulsatile Shear Stress Treatment in Type 2 Diabetes. Biomedicines. 2023; 11(10):2596. https://doi.org/10.3390/biomedicines11102596
Chicago/Turabian StyleUryash, Arkady, Jordan Umlas, Alfredo Mijares, Jose A. Adams, and Jose R. Lopez. 2023. "Enhancing Muscle Intracellular Ca2+ Homeostasis and Glucose Uptake: Passive Pulsatile Shear Stress Treatment in Type 2 Diabetes" Biomedicines 11, no. 10: 2596. https://doi.org/10.3390/biomedicines11102596