

ClC-1 Chloride Channel: Inputs on the Structure–Function Relationship of Myotonia Congenita-Causing Mutations

Abstract
:1. Introduction
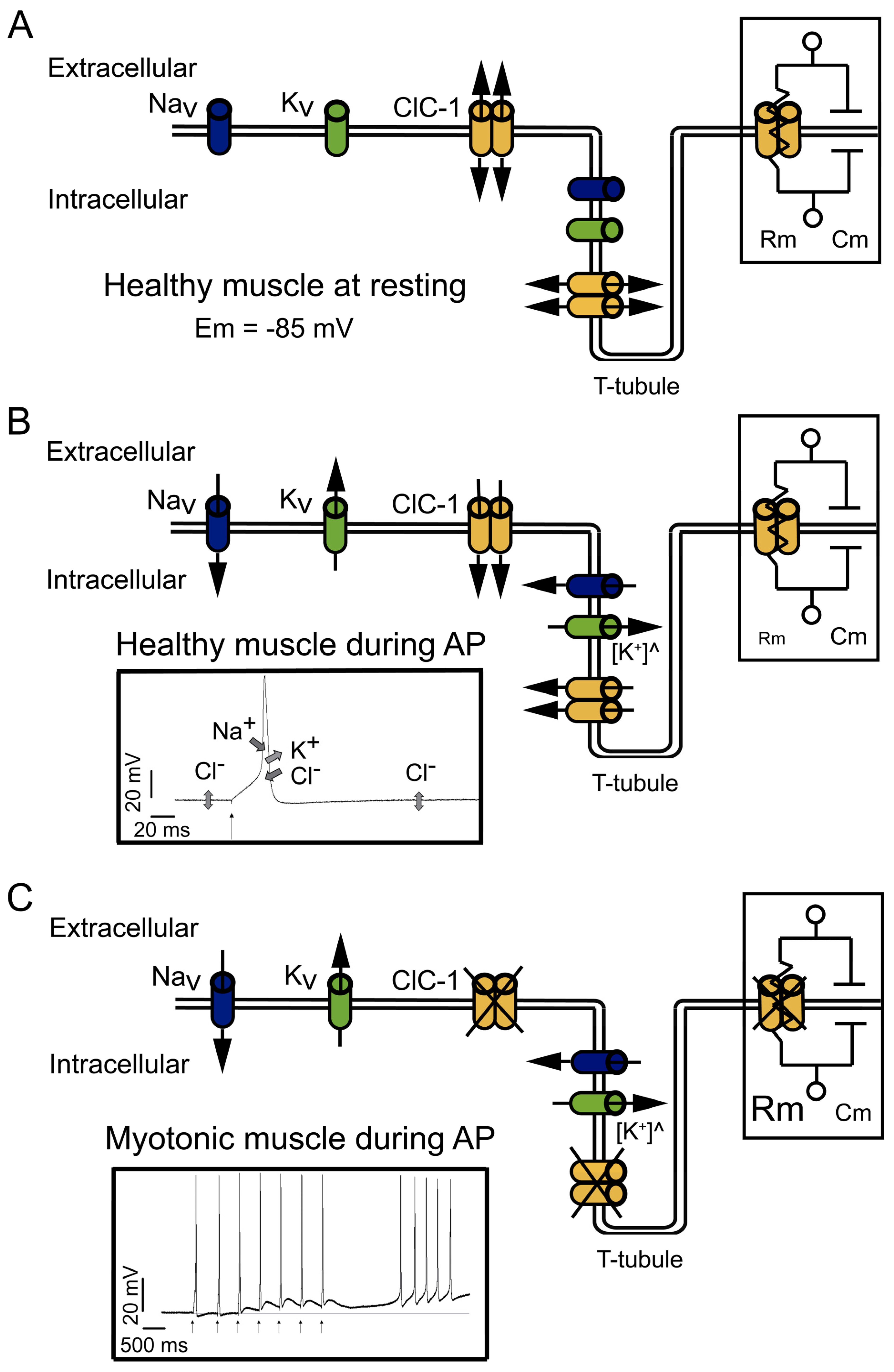
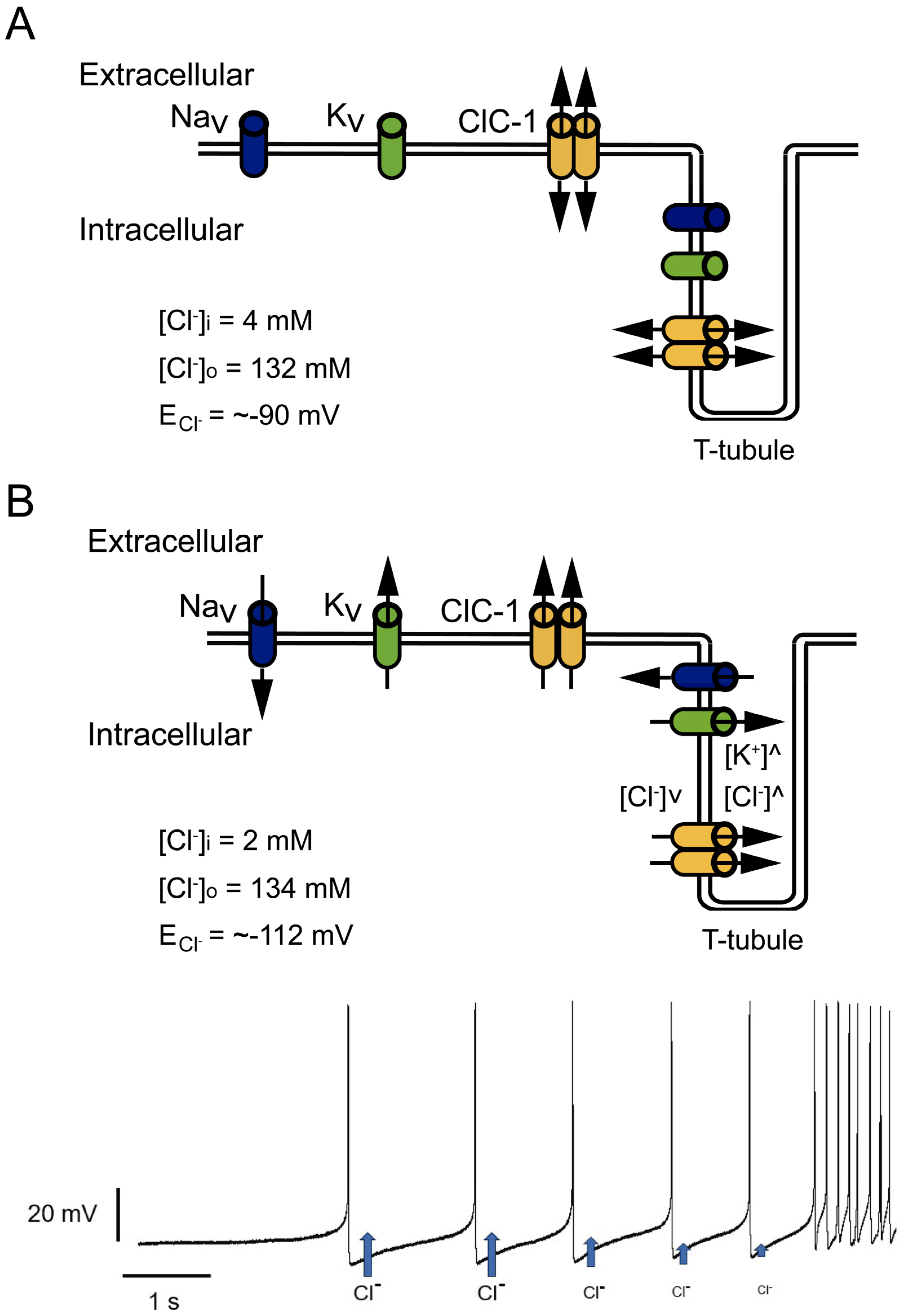
2. Relevance of the ClC-1 Channel in Skeletal Muscle Physiology
3. ClC-1 Localization Controversy: T-Tubule System and/or Sarcolemma
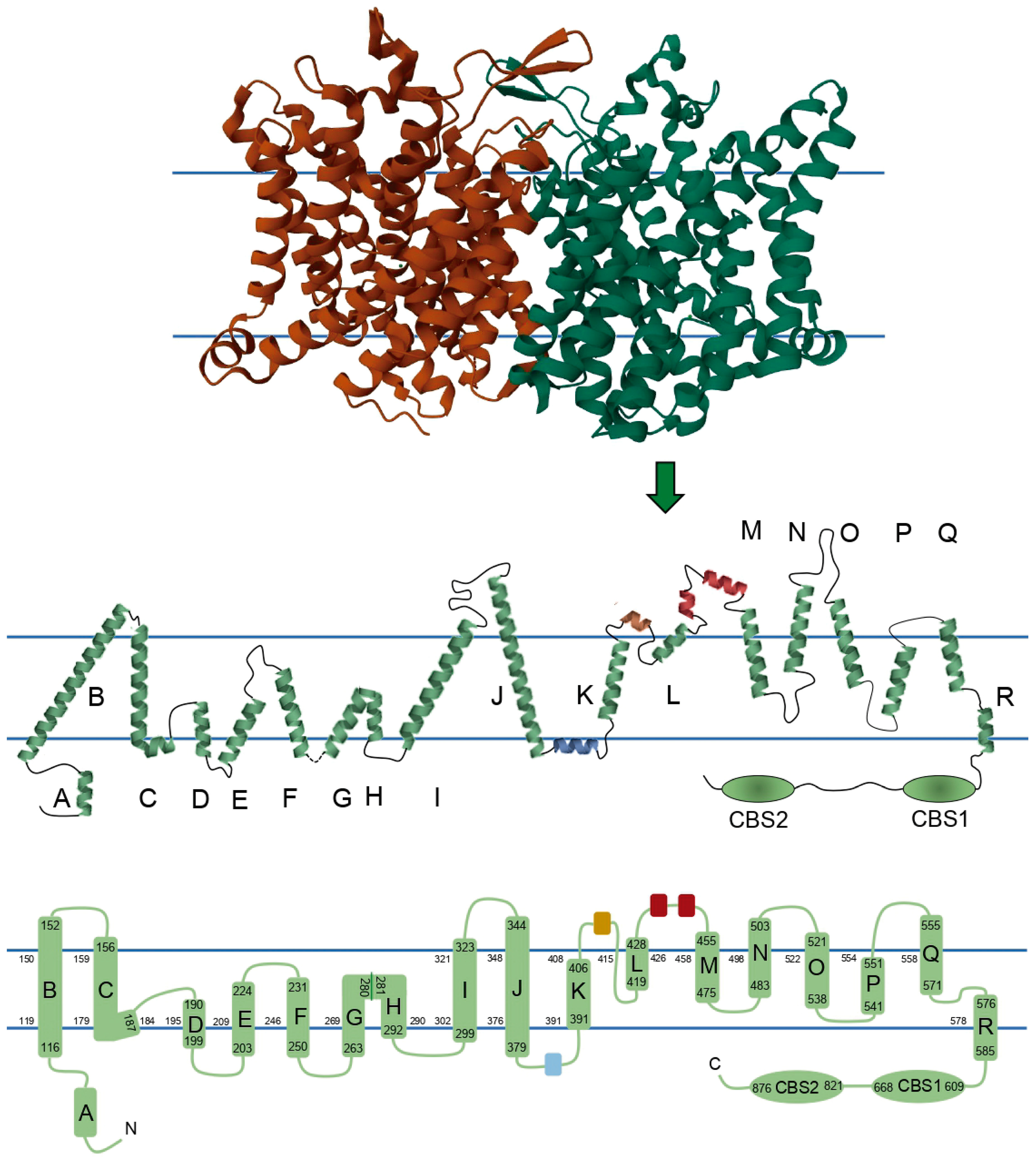
4. ClC-1 Structure–Function Relationship Overview
4.1. Channel Disruption
4.2. Membrane Localization
4.3. Pore Properties
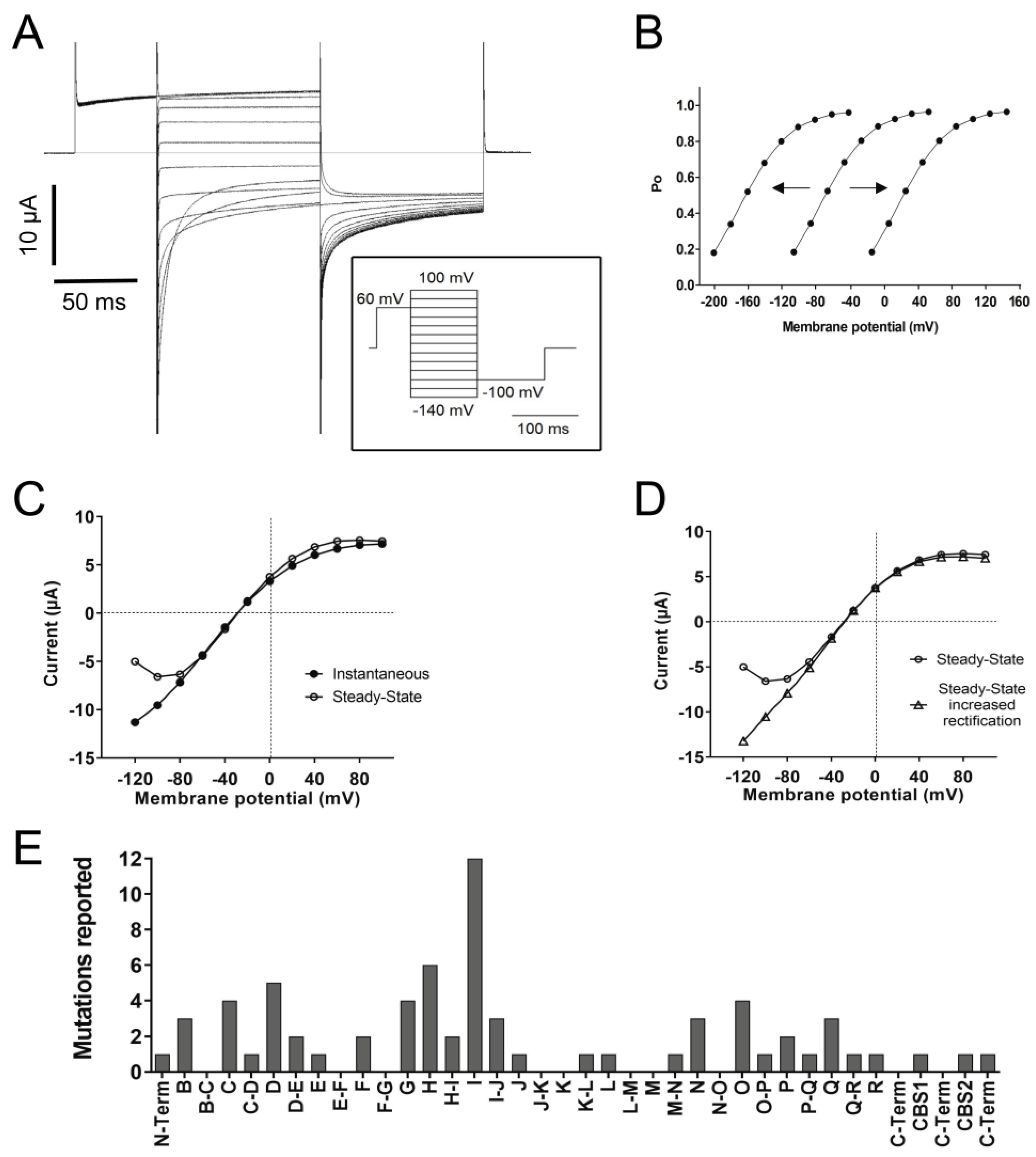
4.4. Gating
4.5. Channel Rectification
4.6. ClC-1 Modulation
4.7. Apparently WT-like Channels
5. Possible Implications for Channel Pharmacological Modulation
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cannon, S.C. Channelopathies of skeletal muscle excitability. Compr. Physiol. 2015, 5, 761–790. [Google Scholar] [PubMed]
- Morales, F.; Pusch, M. An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Channelopathies. Front. Neurol. 2020, 10, 1404. [Google Scholar] [CrossRef]
- Heatwole, C.R.; Moxley, R.T., 3rd. The nondystrophic myotonias. Neurotherapeutics 2007, 4, 238–251. [Google Scholar] [CrossRef]
- Platt, D.; Griggs, R. Skeletal muscle channelopathies: New insights into the periodic paralyses and nondystrophic myotonias. Curr. Opin. Neurol. 2009, 22, 524–531. [Google Scholar] [CrossRef]
- Heatwole, C.R.; Statland, J.M.; Logigian, E.L. The diagnosis and treatment of myotonic disorders. Muscle Nerve 2013, 47, 632–648. [Google Scholar] [CrossRef] [PubMed]
- Soltanzadeh, P. Myotonic Dystrophies: A Genetic Overview. Genes 2022, 13, 367. [Google Scholar] [CrossRef]
- Koch, M.C.; Steinmeyer, K.; Lorenz, C.; Ricker, K.; Wolf, F.; Otto, M.; Zoll, B.; Lehmann-Horn, F.; Grzeschik, K.-H.; Jentsch, T.J. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 1992, 257, 797–800. [Google Scholar] [CrossRef]
- Matthews, E.; Fialho, D.; Tan, S.V.; Venance, S.L.; Cannon, S.C.; Sternberg, D.; Fontaine, B.; Amato, A.A.; Barohn, R.J.; Griggs, R.C.; et al. The non-dystrophic myotonias: Molecular pathogenesis, diagnosis and treatment. Brain 2010, 133, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.E. Myotonia Congenita and Syndromes Associated with Myotonia; Thieme: Sttutgart, Germany, 1977. [Google Scholar]
- Thomsen, J. Tonische Krampfe in willkurlich beweglichen Muskeln in Folge von ererbterpsychischer Disposition (Ataxia muscularis?). Arch. Psychiatr. Nervenkr. 1876, 6, 702–718. [Google Scholar] [CrossRef]
- Sun, C.; Tranebjaerg, L.; Torbergsen, T.; Holmgren, G.; Van Ghelue, M. Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia. Eur. J. Hum. Genet. 2001, 9, 903–909. [Google Scholar] [CrossRef]
- Jurkat-Rott, K.; Lerche, H.; Lehmann-Horn, F. Skeletal muscle channelopathies. J. Neurol. 2002, 249, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.C.; Ricker, K.; Otto, M.; Wolf, F.; Zoll, B.; Lorenz, C.; Jentsch, T.J. Evidence for genetic homogeneity in autosomal recessive generalized myotonia (Becker). J. Med. Genet. 1993, 30, 914–917. [Google Scholar] [CrossRef]
- Ricker, K.; Hertel, G.; Langscheid, K.; Stodieck, G. Myotonia not aggravated by cooling. Force and relaxation of the adductor pollicis in normal subjects and in myotonia as compared to paramyotonia. J. Neurol. 1977, 216, 9–20. [Google Scholar] [CrossRef]
- Park, E.; MacKinnon, R. Structure of the ClC-1 chloride channel from Homo sapiens. Elife 2018, 7, e36629. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Grønberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218. [Google Scholar] [CrossRef]
- Altamura, C.; Desaphy, J.F.; Conte, D.; De Luca, A.; Imbrici, P. Skeletal muscle ClC-1 chloride channels in health and diseases. Pflug. Arch. 2020, 472, 961–975. [Google Scholar] [CrossRef]
- Brenes, O.; Barbieri, R.; Vásquez, M.; Vindas-Smith, R.; Roig, J.; Romero, A.; Morales, F. Functional and Structural Characterization of ClC-1 and Nav1.4 Channels Resulting from CLCN1 and SCN4A Mutations Identified Alone and Coexisting in Myotonic Patients. Cells 2021, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Estevez, R.; Jentsch, T.J. CLC chloride channels: Correlating structure with function. Curr. Opin. Struct. Biol. 2002, 12, 531–539. [Google Scholar] [CrossRef]
- Imbrici, P.; Altamura, C.; Camerino, G.M.; Mangiatordi, G.F.; Conte, E.; Maggi, L.; Camerino, D.C. Multidisciplinary study of a new ClC-1 mutation causing myotonia congenita: A paradigm to understand and treat ion channelopathies. FASEB J. 2016, 30, 3285–3295. [Google Scholar] [CrossRef]
- Imbrici, P.; Altamura, C.; Pessia, M.; Mantegazza, R.; Desaphy, J.F.; Camerino, D.C. ClC-1 chloride channels: State-of-the-art research and future challenges. Front. Cell. Neurosci. 2015, 9, 156. [Google Scholar] [CrossRef]
- Seong, J.Y.; Ha, K.; Hong, C.; Myeong, J.; Lim, H.-H.; Yang, D.; So, I. Helix O modulates voltage dependency of ClC-1. Pflug. Arch. 2017, 469, 183–193. [Google Scholar] [CrossRef]
- Tseng, P.Y.; Yu, W.P.; Liu, H.Y.; Zhang, X.D.; Zou, X.; Chen, T.Y. Binding of ATP to the CBS domains in the C-terminal region of ClC-1. J. Gen. Physiol. 2011, 137, 357–368. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer: Sunderland, MA, USA, 2001. [Google Scholar]
- Kandel, E.R.; Koester, J.D.; Mack, S.H.; Siegelbaum, S. Principles of Natural Science, 6th ed.; McGraw Hill: New York, NY, USA, 2021. [Google Scholar]
- Afzali, A.M.; Ruck, T.; Herrmann, A.M.; Iking, J.; Sommer, C.; Kleinschnitz, C.; Meuth, S.G. The potassium channels TASK2 and TREK1 regulate functional differentiation of murine skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2016, 311, C583–C595. [Google Scholar] [CrossRef]
- Stefani, E.; Chiarandini, D.J. Ionic channels in skeletal muscle. Annu. Rev. Physiol. 1982, 44, 357–372. [Google Scholar] [CrossRef]
- Stolting, G.; Fischer, M.; Fahlke, C. CLC channel function and dysfunction in health and disease. Front. Physiol. 2014, 5, 378. [Google Scholar]
- Zifarelli, G.; Pusch, M. CLC chloride channels and transporters: A biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 2007, 158, 23–76. [Google Scholar]
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef]
- Ruff, R.L.; Lennon, V.A. How myasthenia gravis alters the safety factor for neuromuscular transmission. J. Neuroimmunol. 2008, 201–202, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J. Neuromuscular junction physiology and pthophysiology. In Myasthenia Gravis and Related Disorders; Kaminski, H.L., Kusner, L.L., Eds.; Springer Nature: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Adrian, R.H.; Bryant, S.H. On the repetitive discharge in myotonic muscle fibres. J. Physiol. 1974, 240, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Boron, W.; Boulpaep, E. Medical Physiology, 3rd ed.; Elsevier: Philadelphia, PA, USA, 2017; p. 1297. [Google Scholar]
- Trip, J.; de Vries, J.; Drost, G.; Ginjaar, H.B.; van Engelen, B.G.; Faber, C.G. Health status in non-dystrophic myotonias: Close relation with pain and fatigue. J. Neurol. 2009, 256, 939–947. [Google Scholar] [CrossRef]
- Bennetts, B.; Parker, M.W.; Cromer, B.A. Inhibition of skeletal muscle ClC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 2007, 282, 32780–32791. [Google Scholar] [CrossRef]
- Tseng, P.Y.; Bennetts, B.; Chen, T.Y. Cytoplasmic ATP inhibition of ClC-1 is enhanced by low pH. J. Gen. Physiol. 2007, 130, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; Horowicz, P. The effect of sudden changes in ionic concentrations on the membrane potential of single muscle fibres. J. Physiol. 1960, 153, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Gurnett, C.A.; Kahl, S.D.; Anderson, R.D.; Campbell, K.P. Absence of the skeletal muscle sarcolemma chloride channel ClC-1 in myotonic mice. J. Biol. Chem. 1995, 270, 9035–9038. [Google Scholar] [CrossRef] [PubMed]
- Dulhunty, A.F. Distribution of potassium and chloride permeability over the surface and T-tubule membranes of mammalian skeletal muscle. J. Membr. Biol. 1979, 45, 293–310. [Google Scholar] [CrossRef]
- Palade, P.T.; Barchi, R.L. Characteristics of the chloride conductance in muscle fibers of the rat diaphragm. J. Gen. Physiol. 1977, 69, 325–342. [Google Scholar] [CrossRef]
- Coonan, J.R.; Lamb, G.D. Effect of transverse-tubular chloride conductance on excitability in skinned skeletal muscle fibres of rat and toad. J. Physiol. 1998, 509 Pt 2, 551–564. [Google Scholar] [CrossRef]
- Dutka, T.L.; Murphy, R.M.; Stephenson, D.G.; Lamb, G.D. Chloride conductance in the transverse tubular system of rat skeletal muscle fibres: Importance in excitation-contraction coupling and fatigue. J. Physiol. 2008, 586, 875–887. [Google Scholar] [CrossRef]
- Papponen, H.; Kaisto, T.; Myllyla, V.V.; Myllyla, R.; Metsikko, K. Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp. Neurol. 2005, 191, 163–173. [Google Scholar] [CrossRef]
- Lueck, J.D.; Rossi, A.E.; Thornton, C.A.; Campbell, K.P.; Dirksen, R.T. Sarcolemmal-restricted localization of functional ClC-1 channels in mouse skeletal muscle. J. Gen. Physiol. 2010, 136, 597–613. [Google Scholar] [CrossRef]
- DiFranco, M.; Herrera, A.; Vergara, J.L. Chloride currents from the transverse tubular system in adult mammalian skeletal muscle fibers. J. Gen. Physiol. 2011, 137, 21–41. [Google Scholar] [CrossRef]
- Lamb, G.D.; Murphy, R.M.; Stephenson, D.G. On the localization of ClC-1 in skeletal muscle fibers. J. Gen. Physiol. 2011, 137, 327–329. [Google Scholar] [CrossRef]
- Pusch, M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002, 19, 423–434. [Google Scholar] [CrossRef]
- Desaphy, J.-F.; Gramegna, G.; Altamura, C.; Dinardo, M.M.; Imbrici, P.; George, A.L.; Modoni, A.; LoMonaco, M.; Camerino, D.C. Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenita presenting with different clinical phenotypes. Exp. Neurol. 2013, 248, 530–540. [Google Scholar] [CrossRef]
- Meng, Y.X.; Zhao, Z.; Shen, H.R.; Bing, Q.; Hu, J. Identification of novel mutations of the CLCN1 gene for myotonia congenital in China. Neurol. Res. 2016, 38, 40–44. [Google Scholar] [CrossRef]
- Meyer-Kleine, C.; Steinmeyer, K.; Ricker, K.; Jentsch, T.J.; Koch, M.C. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am. J. Hum. Genet. 1995, 57, 1325–1334. [Google Scholar]
- Macías, M.J.; Teijido, O.; Zifarelli, G.; Martin, P.; Ramirez-Espain, X.; Zorzano, A.; Palacín, M.; Pusch, M.; Estévez, R. Myotonia-related mutations in the distal C-terminus of ClC-1 and ClC-0 chloride channels affect the structure of a poly-proline helix. Biochem. J. 2007, 403, 79–87. [Google Scholar] [CrossRef]
- Papponen, H.; Nissinen, M.; Kaisto, T.; Myllyla, V.V.; Myllyla, R.; Metsikko, K. F413C and A531V but not R894X myotonia congenita mutations cause defective endoplasmic reticulum export of the muscle-specific chloride channel ClC-1. Muscle Nerve 2008, 37, 317–325. [Google Scholar] [CrossRef]
- Hebeisen, S.; Fahlke, C. Carboxy-terminal truncations modify the outer pore vestibule of muscle chloride channels. Biophys J. 2005, 89, 1710–1720. [Google Scholar] [CrossRef]
- Altamura, C.; Ivanova, E.A.; Imbrici, P.; Conte, E.; Camerino, G.M.; Dadali, E.L.; Desaphy, J.F. Pathomechanisms of a CLCN1 Mutation Found in a Russian Family Suffering From Becker’s Myotonia. Front. Neurol. 2020, 11, 1019. [Google Scholar] [CrossRef]
- Gaitan-Penas, H.; Armand-Ugon, M.; Macaya, A.; Estevez, R. CLCN1 Myotonia congenita mutation with a variable pattern of inheritance suggests a novel mechanism of dominant myotonia. Muscle Nerve 2018, 58, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Ronstedt, K.; Sternberg, D.; Detro-Dassen, S.; Gramkow, T.; Begemann, B.; Becher, T.; Kilian, P.; Grieschat, M.; Machtens, J.-P.; Schmalzing, G.; et al. Impaired surface membrane insertion of homo- and heterodimeric human muscle chloride channels carrying amino-terminal myotonia-causing mutations. Sci. Rep. 2015, 5, 15382. [Google Scholar] [CrossRef] [PubMed]
- Simpson, B.J.; Height, T.A.; Rychkov, G.Y.; Nowak, K.J.; Laing, N.G.; Hughes, B.P.; Bretag, A.H. Characterization of three myotonia-associated mutations of the CLCN1 chloride channel gene via heterologous expression. Hum. Mutat. 2004, 24, 185. [Google Scholar] [CrossRef] [PubMed]
- Vindas-Smith, R.; Fiore, M.; Vásquez, M.; Cuenca, P.; del Valle, G.; Lagostena, L.; Gaitán-Peñas, H.; Estevez, R.; Pusch, M.; Morales, F. Identification and Functional Characterization of CLCN1 Mutations Found in Nondystrophic Myotonia Patients. Hum. Mutat. 2016, 37, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sanguinetti, M.C.; Kwiecinski, H.; Ptacek, L.J. Mechanism of inverted activation of ClC-1 channels caused by a novel myotonia congenita mutation. J. Biol. Chem. 2000, 275, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Jeng, C.-J.; Fu, S.-J.; You, C.-Y.; Peng, Y.-J.; Hsiao, C.-T.; Chen, T.-Y.; Tang, C.-Y. Defective Gating and Proteostasis of Human ClC-1 Chloride Channel: Molecular Pathophysiology of Myotonia Congenita. Front. Neurol. 2020, 11, 76. [Google Scholar] [CrossRef]
- Beck, C.L.; Fahlke, C.; George, A.L., Jr. Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proc. Natl. Acad. Sci. USA 1996, 93, 11248–11252. [Google Scholar] [CrossRef]
- Grunnet, M.; Jespersen, T.; Colding-Jørgensen, E.; Schwartz, M.; Klaerke, D.A.; Vissing, J.; Olesen, S.-P.; Dunø, M. Characterization of two new dominant ClC-1 channel mutations associated with myotonia. Muscle Nerve 2003, 28, 722–732. [Google Scholar] [CrossRef]
- Ulzi, G.; Lecchi, M.; Sansone, V.; Redaelli, E.; Corti, E.; Saccomanno, D.; Pagliarani, S.; Corti, S.; Magri, F.; Raimondi, M.; et al. Myotonia congenita: Novel mutations in CLCN1 gene and functional characterizations in Italian patients. J. Neurol. Sci. 2012, 318, 65–71. [Google Scholar] [CrossRef]
- Weinberger, S.; Wojciechowski, D.; Sternberg, D.; Lehmann-Horn, F.; Jurkat-Rott, K.; Becher, T.; Begemann, B.; Fahlke, C.; Fischer, M. Disease-causing mutations C277R and C277Y modify gating of human ClC-1 chloride channels in myotonia congenita. J. Physiol. 2012, 590, 3449–3464. [Google Scholar] [CrossRef]
- Fialho, D.; Schorge, S.; Pucovska, U.; Davies, N.P.; Labrum, R.; Haworth, A.; Stanley, E.; Sud, R.; Wakeling, W.; Davis, M.B.; et al. Chloride channel myotonia: Exon 8 hotspot for dominant-negative interactions. Brain 2007, 130, 3265–3274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Tang, D.; Huang, H.; Tang, H.; Yang, Y.; Yang, M.; Luo, Y.; Tao, H.; Tang, J.; Zhou, X.; et al. Myotonia congenita and periodic hypokalemia paralysis in a consanguineous marriage pedigree: Coexistence of a novel CLCN1 mutation and an SCN4A mutation. PLoS ONE 2020, 15, e0233017. [Google Scholar] [CrossRef] [PubMed]
- Pusch, M.; Steinmeyer, K.; Jentsch, T.J. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 1994, 66, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Saviane, C.; Conti, F.; Pusch, M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999, 113, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Wollnik, B.; Kubisch, C.; Steinmeyer, K.; Pusch, M. Identification of functionally important regions of the muscular chloride channel CIC-1 by analysis of recessive and dominant myotonic mutations. Hum. Mol. Genet. 1997, 6, 805–811. [Google Scholar] [CrossRef]
- Miller, C. Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1982, 299, 401–411. [Google Scholar]
- Accardi, A.; Pusch, M. Fast and slow gating relaxations in the muscle chloride channel ClC-1. J. Gen. Physiol. 2000, 116, 433–444. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Yan, N. Structure of the human voltage-gated sodium channel Na(v)1.4 in complex with beta1. Science 2018, 362, eaau2486. [Google Scholar] [CrossRef]
- Pedersen, T.H.; Riisager, A.; de Paoli, F.V.; Chen, T.Y.; Nielsen, O.B. Role of physiological ClC-1 Cl- ion channel regulation for the excitability and function of working skeletal muscle. J. Gen. Physiol. 2016, 147, 291–308. [Google Scholar] [CrossRef]
- Pusch, M.; Ludewig, U.; Rehfeldt, A.; Jentsch, T.J. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 1995, 373, 527–531. [Google Scholar] [CrossRef]
- Lucchiari, S.; Ulzi, G.; Magri, F.; Bucchia, M.; Corbetta, F.; Servida, M.; Moggio, M.; Comi, G.; Lecchi, M. Clinical evaluation and cellular electrophysiology of a recessive CLCN1 patient. J. Physiol. Pharmacol 2013, 64, 669–678. [Google Scholar] [PubMed]
- Portaro, S.; Altamura, C.; Licata, N.; Camerino, G.M.; Imbrici, P.; Musumeci, O.; Desaphy, J.F. Clinical, Molecular, and Functional Characterization of CLCN1 Mutations in Three Families with Recessive Myotonia Congenita. Neuromolecular Med. 2015, 17, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Fahlke, C.; Rudel, R.; Mitrovic, N.; Zhou, M.; George, A.L., Jr. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron 1995, 15, 463–472. [Google Scholar] [CrossRef]
- Wu, F.; Ryan, A.; Devaney, J.; Warnstedt, M.; Korade-Mirnics, Z.; Poser, B.; Escriva, M.J.; Pegoraro, E.; Yee, A.S.; Felice, K.J.; et al. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain 2002, 125, 2392–2407. [Google Scholar] [CrossRef]
- Burgunder, J.M.; Huifang, S.; Beguin, P.; Baur, R.; Eng, C.S.; Seet, R.C.; Sigel, E. Novel chloride channel mutations leading to mild myotonia among Chinese. Neuromuscul. Disord. 2008, 18, 633–640. [Google Scholar] [CrossRef]
- Bennetts, B.; Yu, Y.; Chen, T.Y.; Parker, M.W. Intracellular beta-nicotinamide adenine dinucleotide inhibits the skeletal muscle ClC-1 chloride channel. J. Biol. Chem. 2012, 287, 25808–25820. [Google Scholar] [CrossRef]
- Suetterlin, K.; Matthews, E.; Sud, R.; McCall, S.; Fialho, D.; Burge, J.; Jayaseelan, D.; Haworth, A.; Sweeney, M.G.; Kullmann, D.M.; et al. Translating genetic and functional data into clinical practice: A series of 223 families with myotonia. Brain 2022, 145, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shelat, A.A.; Guy, R.K.; Gopinath, V.S.; Ma, T.; Du, K.; Verkman, A.S. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J. Biol. Chem. 2003, 278, 35079–35085. [Google Scholar] [CrossRef]
- Liu, J.; Bihler, H.; Farinha, C.M.; Awatade, N.T.; Romão, A.M.; Mercadante, D.; Cheng, Y.; Musisi, I.; Jantarajit, W.; Wang, Y.; et al. Partial rescue of F508del-cystic fibrosis transmembrane conductance regulator channel gating with modest improvement of protein processing, but not stability, by a dual-acting small molecule. Br. J. Pharmacol. 2018, 175, 1017–1038. [Google Scholar] [CrossRef]
- Chen, P.-C.; Olson, E.M.; Zhou, Q.; Kryukova, Y.; Sampson, H.M.; Thomas, D.Y.; Shyng, S.-L. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J. Biol. Chem. 2013, 288, 20942–20954. [Google Scholar] [CrossRef]
- Pedersen, T.H.; Macdonald, W.A.; Broch-Lips, M.; Halldorsdottir, O.; Bækgaard Nielsen, O. Chloride channel inhibition improves neuromuscular function under conditions mimicking neuromuscular disorders. Acta Physiol. 2021, 233, e13690. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Guzman, L.; Jimenez-Huezo, G.; Arguedas, A.; Leal, A. Mutational survivorship bias: The case of PNKP. PLoS ONE 2020, 15, e0237682. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Function | Mutations Reported |
|---|---|---|
| N-terminal and A | Membrane localization + Voltage-dependence of open probability + Link between the two subunits * | 17 |
| B | Membrane localization + Voltage-dependence of fast and slow gates + Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + Effects on rectification + Link between the two subunits * | 15 |
| B-C loop | No mutations were reported = unknown function | 0 |
| C | Membrane localization + Affects ion selectivity + Voltage-dependence of open probability + Deactivation at negative voltages of fast and slow gates + | 11 |
| C-D loop | Voltage-dependence of fast and slow gates + Link C-D is a loop that opens the vestibule, providing intracellular access * Directly involved in anion binding * | 5 |
| D | Contribute to the ClC-1 ion pathway + Part of the selectivity filter + Voltage-dependence of fast and slow gates + Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + Effects on rectification + Directly involved in anion binding * | 10 |
| D-E loop | NAD+ inhibition + Voltage-dependence of open probability + | 2 |
| E | Voltage-dependence of open probability + | 9 |
| E-F loop | Effect on current amplitude = specific function unknown | 3 |
| F | Voltage-dependence of open probability + Contribute to the ClC-1 ion pathway * Part of the selectivity filter * Directly involved in anion binding * Part of the fast gate (E232) * | 11 |
| F-G loop | NAD+ inhibition + | ~5 |
| G | Effects on ion selectivity and single channel conductance, suggesting a role in pore functioning + Effects on rectification + Voltage-dependence of fast and slow gating + Deactivation at negative voltages of fast and slow gates + Membrane localization + | 12 |
| H | Voltage-dependence of open probability + Part of the slow gate + Effects on rectification + Link between the two subunits * H-P interaction is probably necessary for channel assembly * Probably interact with CBS2 regarding their phosphorylation status during PKC modulation * | 17 |
| H-I loop | Voltage-dependence of open probability + Deactivation at negative voltages of slow gate + Effects on rectification + | 3 |
| I | Affects ion selectivity + Voltage-dependence of open probability + Link between the two subunits but not reported effects on slow gate kinetics * Probably interact with CBS2 regarding their phosphorylation status during PKC modulation * | 22 |
| I-J loop | Voltage-dependence of open probability + Probably part of the slow gate + Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + | 19 |
| J | Voltage-dependence of fast and slow gating + Conformational stability of the channel * | 11 |
| Helical stretch J-K | CBS interaction * | 0 |
| K | Poorly analyzed mutations reported = unknown function | 7 |
| Helical stretch K-L | Membrane localization + Voltage-dependence of open probability + | 7 |
| L | Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + | 10 |
| Helical stretch L-M | Voltage-dependence of open probability + Involved in trafficking + Deactivation at negative voltages of fast and slow gates + | 7 |
| M | Poorly analyzed mutations reported = unknown function | 8 |
| M-N loop | Voltage-dependence of the slow gate + Affects ion selectivity + Deactivation at negative voltages of fast and slow gates + | 11 |
| N | Voltage-dependence of fast and slow gates + Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + Contribute to the ClC-1 ion pathway + Part of the selectivity filter + Effects on rectification + Membrane localization + Directly involved in anion binding * | 14 |
| N-O loop | No analyzed mutations were reported = unknown function | 1 |
| O | Voltage-dependence of fast and slow gates + Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + Effects on rectification + Membrane localization + Conformational stability of the channel * Modulation of voltage dependency by pH * | 14 |
| O-P loop | Probably part of the slow gate + | 1 |
| P | Part of the slow gate + Link between the two subunits * H-P interaction is probably necessary for channel assembly * Role in binding zinc (possibly through the slow gate) * | 5 |
| P-Q loop | Voltage-dependence of open probability + Deactivation at negative voltages of the slow gate + Effects on rectification + | 2 |
| Q | Part of the slow gate + Voltage-dependence of fast and slow gates + Channel opening kinetics at positive voltages and deactivation at negative voltages of fast and slow gates + Effects on rectification + Link between the two subunits * | 10 |
| Q-R loop | Voltage-dependence of open probability + | 2 |
| R | Voltage-dependence of open probability + Contribute to the ClC-1 ion pathway * Part of the selectivity filter * Directly involved in anion binding * Helix R is close to helix A of the other monomer, possibly suggesting an interaction that might be important for slow gating * | 1 |
| C-terminal loops | Voltage-dependence of fast and slow gating + Involved in trafficking + Possibly involved in the pore structure + Modulation of the slow gate + ATP binding site * PKC phosphorylation site * Interacts with helix R linker, helix D, and intracellular H-I loop * | 4 35 12 |
| CBS1 CBS2 | Modulation of the slow gate + Channel opening kinetics at positive voltages + Involved in trafficking * Involved in oligomerization * | 20 19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenes, O.; Pusch, M.; Morales, F. ClC-1 Chloride Channel: Inputs on the Structure–Function Relationship of Myotonia Congenita-Causing Mutations. Biomedicines 2023, 11, 2622. https://doi.org/10.3390/biomedicines11102622
Brenes O, Pusch M, Morales F. ClC-1 Chloride Channel: Inputs on the Structure–Function Relationship of Myotonia Congenita-Causing Mutations. Biomedicines. 2023; 11(10):2622. https://doi.org/10.3390/biomedicines11102622
Chicago/Turabian StyleBrenes, Oscar, Michael Pusch, and Fernando Morales. 2023. "ClC-1 Chloride Channel: Inputs on the Structure–Function Relationship of Myotonia Congenita-Causing Mutations" Biomedicines 11, no. 10: 2622. https://doi.org/10.3390/biomedicines11102622
APA StyleBrenes, O., Pusch, M., & Morales, F. (2023). ClC-1 Chloride Channel: Inputs on the Structure–Function Relationship of Myotonia Congenita-Causing Mutations. Biomedicines, 11(10), 2622. https://doi.org/10.3390/biomedicines11102622