
The Expression of the Alpha7 Nicotinic Acetylcholine Receptor and the Effect of Smoking in Curdlan-Administered SKG Mice
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Induction of Arthritis and Ankylosis in SKG Mice
2.2. Cigarette Smoke Exposure
2.3. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) for Metabolomic Analysis
2.4. Clinical Scoring of Peripheral Arthritis
2.5. Histological Analysis
2.6. Immunofluorescence Staining
2.7. Imaging with a Fluorescent In Vivo Bisphosphonate Agent
2.8. Statistical Analysis and Schematic Diagrams
3. Results
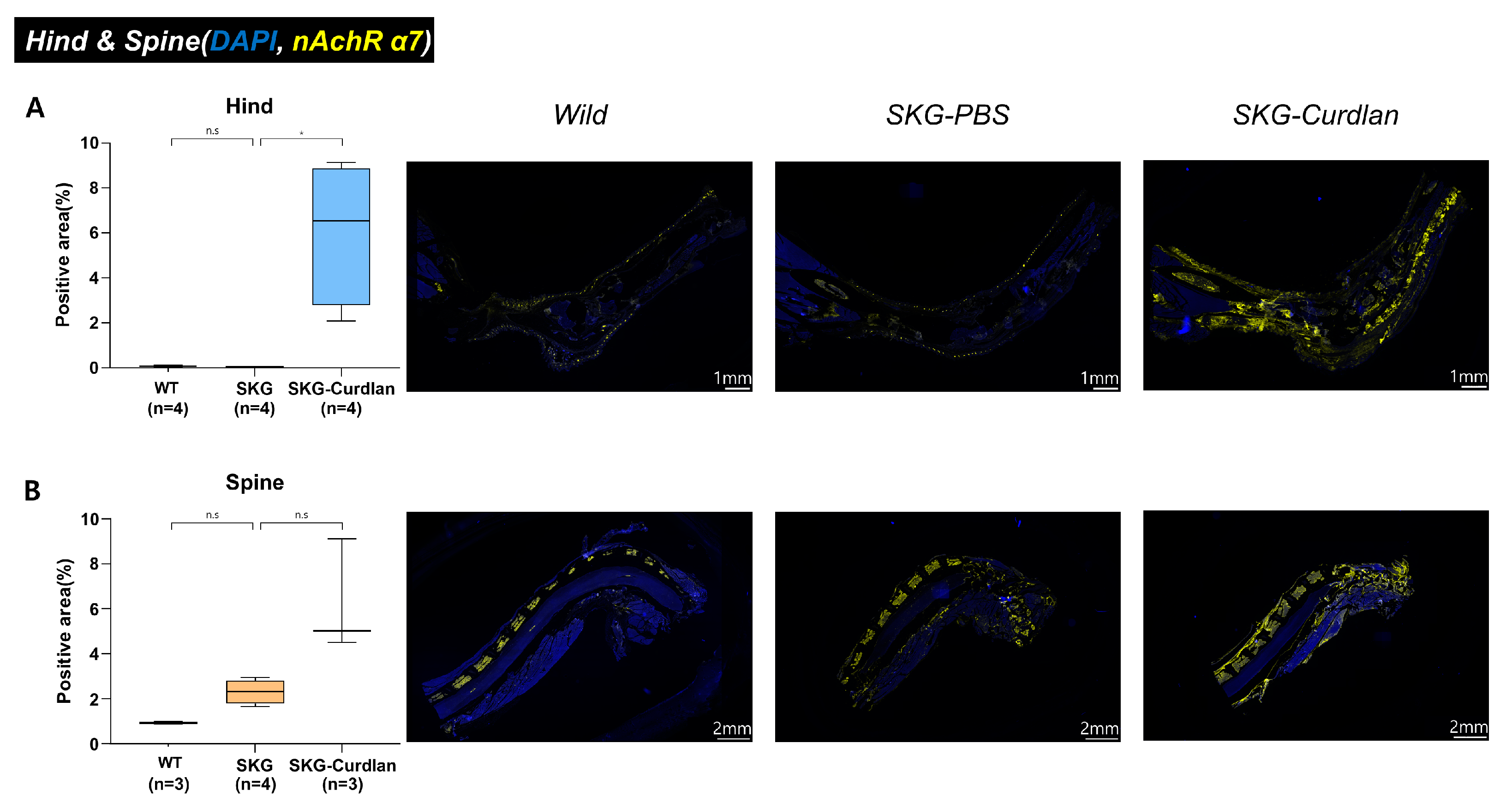
3.1. Expression of α7 nAChR in the Joint Tissue of Curdlan-Administered SKG Mice
3.2. Effect of Cigarette Smoke on Peripheral Arthritis
3.3. Histologic Examination of Peripheral Joints
3.4. Osteoblast Activity in the Spine
3.5. Effect of Cigarette Smoke on the Expression of α7 nAChRs, IL-17A, FOXP3, and F4/80 in the Synovium
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Heijde, D.; Ramiro, S.; Landewé, R.; Baraliakos, X.; Van den Bosch, F.; Sepriano, A.; Regel, A.; Ciurea, A.; Dagfinrud, H.; Dougados, M.; et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; Braun, J.; Dougados, M.; Baeten, D. Axial spondyloarthritis. Nat. Rev. Dis. Prim. 2015, 1, 15013. [Google Scholar] [CrossRef] [PubMed]
- Gaston, J.S.H.; Jadon, D.R. Th17 cell responses in spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 777–796. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, W.B.; McInnes, I.B. Th17 cells and IL-17 A—Focus on immunopathogenesis and immunotherapeutics. Semin. Arthritis Rheum. 2013, 43, 158–170. [Google Scholar] [CrossRef]
- Mei, Y.; Pan, F.; Gao, J.; Ge, R.; Duan, Z.; Zeng, Z.; Liao, F.; Xia, G.; Wang, S.; Xu, S.; et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin. Rheumatol. 2011, 30, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, V.; Gracey, E.; Brown, M.A.; Inman, R.D.; Haroon, N. Pathogenesis of ankylosing spondylitis—Recent advances and future directions. Nat. Rev. Rheumatol. 2017, 13, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ regulatory T cells and their functional regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Liao, H.; Chen, C.; Chen, W.; Wang, H.; Su, K. The Clinical Application of Anti-CCP in Rheumatoid Arthritis and Other Rheumatic Diseases. Biomark. Insights 2007, 2, 117727190700200. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chen, H.-A.; Lu, C.-L.; Liao, H.-T.; Liu, C.-H.; Tsai, C.-Y.; Chou, C.-T. Association of cigarette smoking with Chinese ankylosing spondylitis patients in Taiwan: A poor disease outcome in systemic inflammation, functional ability, and physical mobility. Clin. Rheumatol. 2013, 32, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, S.S.; Mydel, P.; Jonsson, I.-M.; Senior, R.M.; Tarkowski, A.; Bokarewa, M. Smoking and nicotine exposure delay development of collagen-induced arthritis in mice. Arthritis Res. Ther. 2009, 11, R88. [Google Scholar] [CrossRef]
- Lv, J.; Ji, X.; Li, Z.; Hao, H. The role of the cholinergic anti-inflammatory pathway in autoimmune rheumatic diseases. Scand. J. Immunol. 2021, 94, e13092. [Google Scholar] [CrossRef]
- Maanen, M.A.V.; Stoof, S.P.; Van Der Zanden, E.P.; De Jonge, W.J.; Janssen, R.A.; Fischer, D.F.; Vandeghinste, N.; Brys, R.; Vervoordeldonk, M.J.; Tak, P.P. The α7 nicotinic acetylcholine receptor on fibroblast-like synoviocytes and in synovial tissue from rheumatoid arthritis patients: A possible role for a key neurotransmitter in synovial inflammation. Arthritis Rheum. 2009, 60, 1272–1281. [Google Scholar] [CrossRef]
- van Maanen, M.A.; Papke, R.L.; Koopman, F.A.; Koepke, J.; Bevaart, L.; Clark, R.; Lamppu, D.; Elbaum, D.; LaRosa, G.J.; Tak, P.P.; et al. Two Novel α7 Nicotinic Acetylcholine Receptor Ligands: In Vitro Properties and Their Efficacy in Collagen-Induced Arthritis in Mice. PLoS ONE 2015, 10, e0116227. [Google Scholar] [CrossRef]
- Van Maanen, M.A.; Lebre, M.C.; Van Der Poll, T.; Larosa, G.J.; Elbaum, D.; Vervoordeldonk, M.J.; Tak, P.P. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009, 60, 114–122. [Google Scholar] [CrossRef]
- Tregellas, J.R.; Wylie, K.P. Alpha7 Nicotinic Receptors as Therapeutic Targets in Schizophrenia. Nicotine Tob. Res. 2019, 21, 349–356. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, H.; Zou, M.; Yuan, Q.; Huang, Z.; Pan, X.; Zhang, W. Nicotine in Inflammatory Diseases: Anti-Inflammatory and Pro-Inflammatory Effects. Front. Immunol. 2022, 13, 826889. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Yang, J.; Xie, R.; Ren, Y.; Fan, H. Regulatory effect of nicotine on collagen-induced arthritis and on the induction and function of in vitro-cultured Th17 cells. Mod. Rheumatol. 2014, 24, 781–787. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, N.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Liu, Z.; Han, B.; Li, P.; Wang, Z.; Fan, Q. Activation of α7nAChR by Nicotine Reduced the Th17 Response in CD4+T Lymphocytes. Immunol. Investig. 2014, 43, 667–674. [Google Scholar] [CrossRef]
- Li, S.; Zhou, B.; Liu, B.; Zhou, Y.; Zhang, H.; Li, T.; Zuo, X. Activation of the cholinergic anti-inflammatory system by nicotine attenuates arthritis via suppression of macrophage migration. Mol. Med. Rep. 2016, 14, 5057–5064. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Jacob, P., III. Metabolism of nicotine to cotinine studied by a dual stable isotope method. Clin. Pharmacol. Ther. 1994, 56, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Rehani, K.; Scott, D.A.; Renaud, D.; Hamza, H.; Williams, L.R.; Wang, H.; Martin, M. Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim. Biophys. Acta 2008, 1783, 375–382. [Google Scholar] [CrossRef]
- Hukkanen, J.; Jacob, P., 3rd; Benowitz, N.L. Metabolism and Disposition Kinetics of Nicotine. Pharmacol. Rev. 2005, 57, 79–115. [Google Scholar] [CrossRef]
- Zhao, S.; Jones, G.T.; Macfarlane, G.J.; Hughes, D.M.; Dean, L.E.; Moots, R.J.; Goodson, N.J. Associations between smoking and extra-axial manifestations and disease severity in axial spondyloarthritis: Results from the BSR Biologics Register for Ankylosing Spondylitis (BSRBR-AS). Rheumatology 2019, 58, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Bessis, N.; Decker, P.; Assier, E.; Semerano, L.; Boissier, M.-C. Arthritis models: Usefulness and interpretation. Semin. Immunopathol. 2017, 39, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Ruutu, M.; Thomas, G.; Steck, R.; Degli-Esposti, M.A.; Zinkernagel, M.S.; Alexander, K.; Velasco, J.; Strutton, G.; Tran, A.; Benham, H.; et al. β-glucan triggers spondylarthritis and Crohn’s disease-like ileitis in SKG mice. Arthritis Rheum. 2012, 64, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Thomas, R. The SKG model of spondyloarthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 895–909. [Google Scholar] [CrossRef]
- Lim, D.-H.; Lee, E.-J.; Kwon, O.C.; Hong, S.; Lee, C.-K.; Yoo, B.; Youn, J.; Kim, T.-H.; Kim, Y.-G. Effect of tumor necrosis factor inhibition on spinal inflammation and spinal ankylosis in SKG mice. Sci. Rep. 2019, 9, 18000. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Rehaume, L.M.; Hasnain, S.Z.; Velasco, J.; Baillet, A.C.; Ruutu, M.; Kikly, K.; Wang, R.; Tseng, H.-W.; Thomas, G.P.; et al. Interleukin-23 Mediates the Intestinal Response to Microbial β-1,3-Glucan and the Development of Spondyloarthritis Pathology in SKG Mice. Arthritis Rheumatol. 2014, 66, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.K.; Lindgaard, L.M.; Wogensen, L.; Dagnæs-Hansen, F.; Thomsen, J.S.; Sakaguchi, S.; Stengaard-Pedersen, K.; Hauge, E.-M. SKG arthritis as a model for evaluating therapies in rheumatoid arthritis with special focus on bone changes. Rheumatol. Int. 2013, 33, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Keith, R.C.; Sokolove, J.; Edelman, B.L.; Lahey, L.; Redente, E.F.; Holers, V.M.; Sakaguchi, S.; Robinson, W.H.; Riches, D.W.H. Brief Report: Testosterone Is Protective in the Sexually Dimorphic Development of Arthritis and Lung Disease in SKG Mice. Arthritis Rheum. 2013, 65, 1487–1493. [Google Scholar] [CrossRef]
- Jang, Y.O.; Lee, S.H.; Choi, J.J.; Kim, D.-H.; Choi, J.-M.; Kang, M.-J.; Oh, Y.-M.; Park, Y.-J.; Shin, Y.; Lee, S.W. Fecal microbial transplantation and a high fiber diet attenuates emphysema development by suppressing inflammation and apoptosis. Exp. Mol. Med. 2020, 52, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Hayer, S.; Vervoordeldonk, M.J.; Denis, M.C.; Armaka, M.; Hoffmann, M.; Bäcklund, J.; Nandakumar, K.S.; Niederreiter, B.; Geka, C.; Fischer, A.; et al. ‘SMASH’ recommendations for standardised microscopic arthritis scoring of histological sections from inflammatory arthritis animal models. Ann. Rheum. Dis. 2021, 80, 714–726. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.S.; Goodson, N.J.; Robertson, S.; Gaffney, K. Smoking in spondyloarthritis: Unravelling the complexities. Rheumatology 2020, 59, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- Videm, V.; Cortes, A.; Thomas, R.; Brown, M.A. Current Smoking is Associated with Incident Ankylosing Spondylitis—The HUNT Population-based Norwegian Health Study. J. Rheumatol. 2014, 41, 2041–2048. [Google Scholar] [CrossRef]
- Zhang, H.; Wan, W.; Liu, J.; Dai, S.; Zou, Y.; Qian, Q.; Ding, Y.; Xu, X.; Ji, H.; He, H.; et al. Smoking quantity determines disease activity and function in Chinese patients with ankylosing spondylitis. Clin. Rheumatol. 2018, 37, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Challoner, B.; Khattak, M.; Moots, R.J.; Goodson, N.J. Increasing smoking intensity is associated with increased disease activity in axial spondyloarthritis. Rheumatol. Int. 2017, 37, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Machado, P.; van der Heijde, D.; D’Agostino, M.-A.; Dougados, M. Smokers in early axial spondyloarthritis have earlier disease onset, more disease activity, inflammation and damage, and poorer function and health-related quality of life: Results from the DESIR cohort. Ann. Rheum. Dis. 2011, 71, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Joseph, F.B.; Edward, R.B.; McKennis, H., Jr. studies on the respiratory and cardiovascular effects of (−)-cotinine. J. Pharmacol. Exp. Ther. 1962, 137, 313. [Google Scholar]
- Edward, R.B.; McKennis, H., Jr. Studies on the metabolism of (−)-cotinine in the human. J. Pharmacol. Exp. Ther. 1962, 135, 306. [Google Scholar]
- Nakamura, A.; Talukdar, A.; Nakamura, S.; Pathan, E.; Haroon, N. Bone formation in axial spondyloarthritis: Is disease modification possible? Best Pract. Res. Clin. Rheumatol. 2019, 33, 101491. [Google Scholar] [CrossRef] [PubMed]
- Aspera-Werz, R.H.; Ehnert, S.; Heid, D.; Zhu, S.; Chen, T.; Braun, B.; Sreekumar, V.; Arnscheidt, C.; Nussler, A.K. Nicotine and Cotinine Inhibit Catalase and Glutathione Reductase Activity Contributing to the Impaired Osteogenesis of SCP-1 Cells Exposed to Cigarette Smoke. Oxid. Med. Cell. Longev. 2018, 2018, 3172480. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Schett, G. Effects of the IL-23–IL-17 pathway on bone in spondyloarthritis. Nat. Rev. Rheumatol. 2018, 14, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Miossec, P. IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect. 2009, 11, 625–630. [Google Scholar] [CrossRef]
- Shen, H.; Goodall, J.C.; Gaston, J.S.H. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 2009, 60, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. The cholinergic anti-inflammatory pathway: Towards innovative treatment of rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Therriault, M.J.; Proulx, L.I.; Castonguay, A.; Bissonnette, É.Y. Immunomodulatory effects of the tobacco-specific carcinogen, NNK, on alveolar macrophages. Clin. Exp. Immunol. 2003, 132, 232–238. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-E.; Lee, J.-H.; Lee, E.-J.; Kim, D.H.; Jeong, M.R.; Hong, S.; Lee, C.-K.; Yoo, B.; Youn, J.; Chang, E.-J.; et al. The Expression of the Alpha7 Nicotinic Acetylcholine Receptor and the Effect of Smoking in Curdlan-Administered SKG Mice. Biomedicines 2023, 11, 2757. https://doi.org/10.3390/biomedicines11102757
Kim Y-E, Lee J-H, Lee E-J, Kim DH, Jeong MR, Hong S, Lee C-K, Yoo B, Youn J, Chang E-J, et al. The Expression of the Alpha7 Nicotinic Acetylcholine Receptor and the Effect of Smoking in Curdlan-Administered SKG Mice. Biomedicines. 2023; 11(10):2757. https://doi.org/10.3390/biomedicines11102757
Chicago/Turabian StyleKim, Young-Eun, Jae-Hyun Lee, Eun-Ju Lee, Do Hoon Kim, Mi Ryeong Jeong, Seokchan Hong, Chang-Keun Lee, Bin Yoo, Jeehee Youn, Eun-Ju Chang, and et al. 2023. "The Expression of the Alpha7 Nicotinic Acetylcholine Receptor and the Effect of Smoking in Curdlan-Administered SKG Mice" Biomedicines 11, no. 10: 2757. https://doi.org/10.3390/biomedicines11102757
APA StyleKim, Y. -E., Lee, J. -H., Lee, E. -J., Kim, D. H., Jeong, M. R., Hong, S., Lee, C. -K., Yoo, B., Youn, J., Chang, E. -J., & Kim, Y. -G. (2023). The Expression of the Alpha7 Nicotinic Acetylcholine Receptor and the Effect of Smoking in Curdlan-Administered SKG Mice. Biomedicines, 11(10), 2757. https://doi.org/10.3390/biomedicines11102757