
A Current Landscape on Alport Syndrome Cases: Characterization, Therapy and Management Perspectives

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
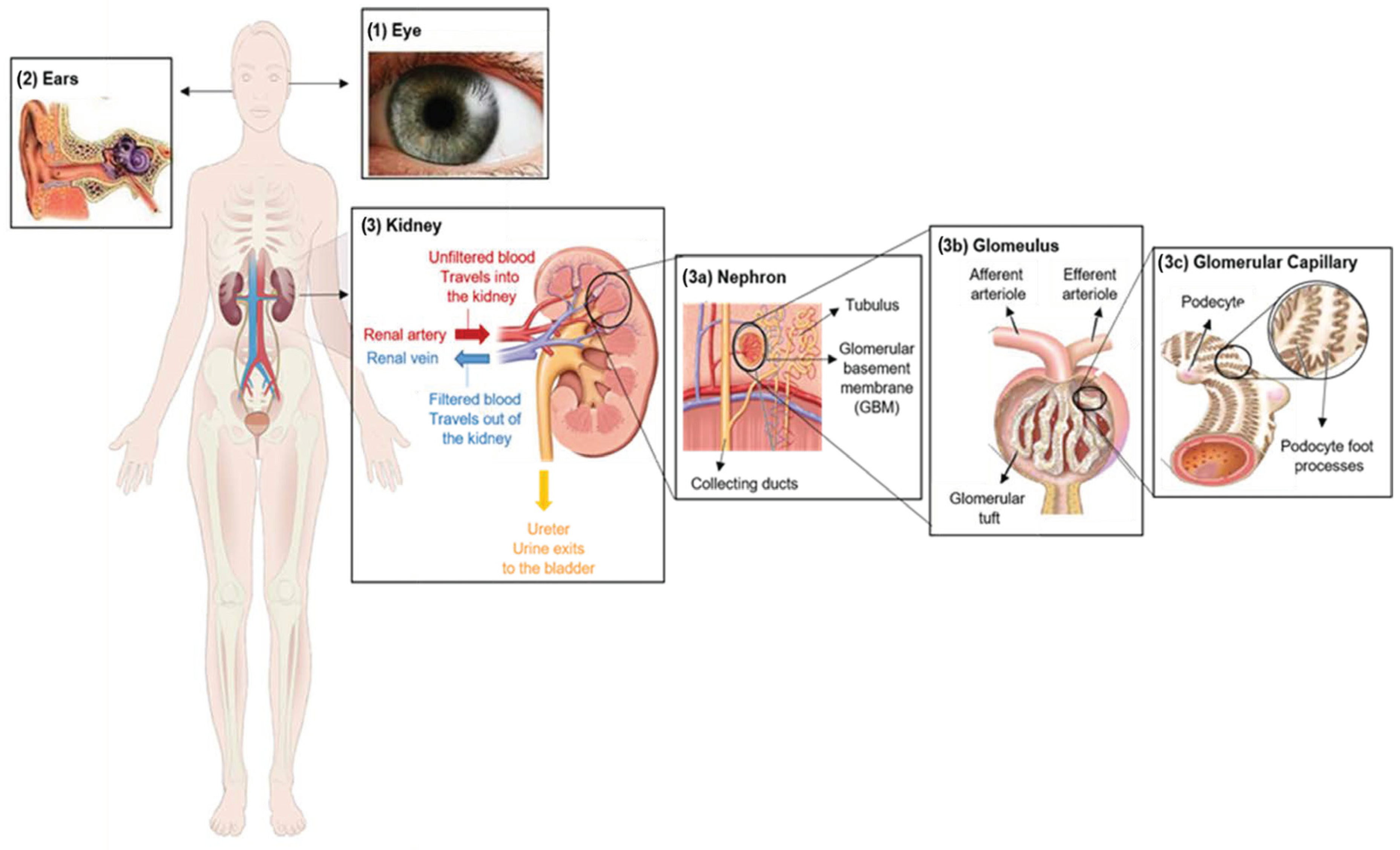
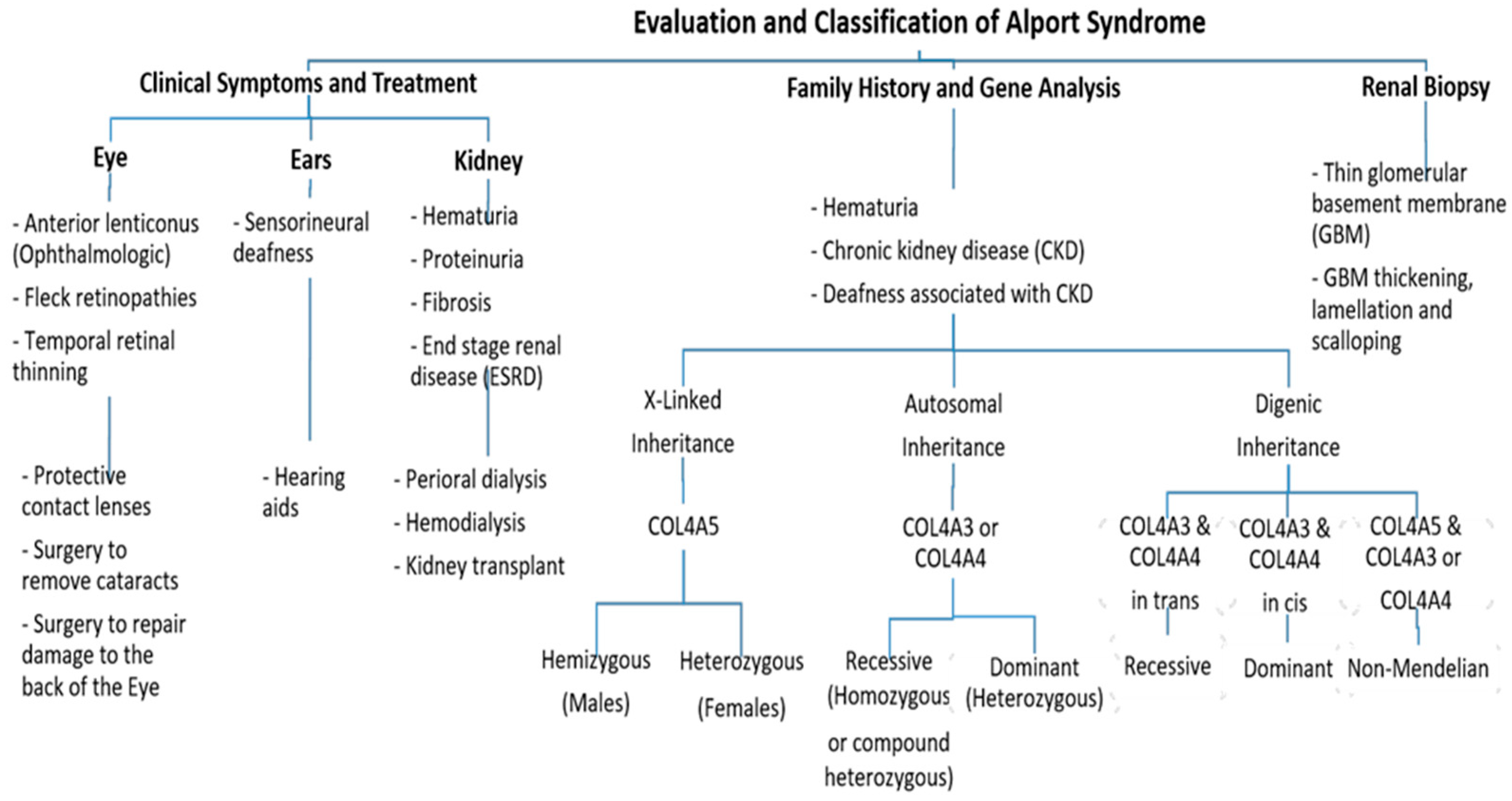
2. Clinical Characterization of Alport Syndrome
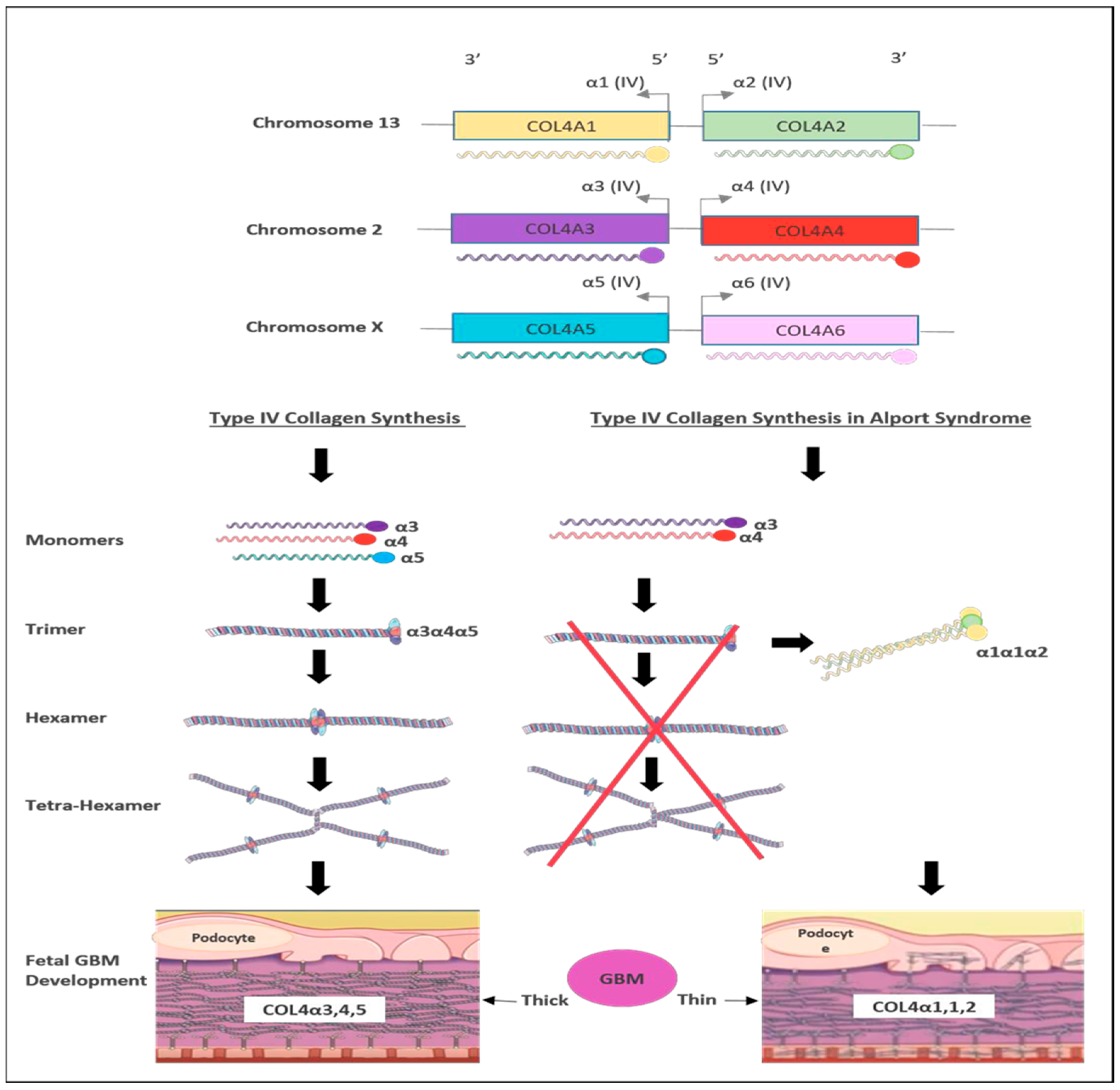
3. Genetics of Alport Syndrome
4. Cases of Alport Syndrome in Saudi Arabia
5. Current Therapies for Alport Syndrome Management
5.1. Chemical Drugs
5.2. Molecular Therapies
6. Gene-Editing-Based Therapies
7. Recommendations for Alport Syndrome Management
7.1. Kidney Transplantation
7.2. Genetic Counseling and Molecular Genetic Testing
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Williamson, D.A. Alport’s syndrome of hereditary nephritis with deafness. Lancet 1961, 278, 1321–1323. [Google Scholar] [CrossRef]
- Flinter, F. Alport’s syndrome. J. Med. Genet. 1997, 34, 326–330. [Google Scholar] [PubMed]
- Watson, S.; Padala, S.A.; Hashmi, M.F.; Bush, J.S. Alport Syndrome; StatPearls: Treasure Island, FL, USA, 2017. [Google Scholar]
- Gibson, J.T.; Huang, M.; Dabrera, M.S.C.; Shukla, K.; Rothe, H.; Hilbert, P.; Deltas, C.; Storey, H.; Lipska-Ziętkiewicz, B.S.; Chan, M.M.Y.; et al. Genotype–phenotype correlations for COL4A3–COL4A5 variants resulting in Gly substitutions in Alport syndrome. Sci. Rep. 2022, 12, 2722. [Google Scholar] [PubMed]
- Gross, O.; Kashtan, C.E. Treatment of Alport syndrome: Beyond animal models. Kidney Int. 2009, 76, 599–603. [Google Scholar] [CrossRef]
- Suh, J.H.; Miner, J.H. The glomerular basement membrane as a barrier to albumin. Nat. Rev. Nephrol. 2013, 9, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2019, 23, 158–168. [Google Scholar]
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport syndrome classification and management. Kidney Med. 2020, 2, 639–649. [Google Scholar] [PubMed]
- Mallett, A.; Tang, W.; Clayton, P.A.; Stevenson, S.; McDonald, S.P.; Hawley, C.M.; Badve, S.V.; Boudville, N.; Brown, F.G.; Campbell, S.B.; et al. End-stage kidney disease due to Alport syndrome: Outcomes in 296 consecutive Australia and New Zealand Dialysis and Transplant Registry cases. Nephrol. Dial. Transplant. 2014, 29, 2277–2286. [Google Scholar]
- Yamamura, T.; Nozu, K.; Fu, X.J.; Nozu, Y.; Ye, M.J.; Shono, A.; Yamanouchi, S.; Minamikawa, S.; Morisada, N.; Nakanishi, K.; et al. Natural history and genotype–phenotype correlation in female X-linked Alport syndrome. Kidney Int. Rep. 2017, 2, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, O.K.; Flinter, F.; et al. X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J. Am. Soc. Nephrol. 2000, 11, 649–657. [Google Scholar] [CrossRef]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610. [Google Scholar] [CrossRef]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Faculty Opinions recommendation of Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.P.; Lemann, J. In hereditary nephritis angiotensin-converting enzyme inhibition decreases proteinuria and may slow the rate of progression. Am. J. Kidney Dis. 1996, 27, 199–203. [Google Scholar] [CrossRef]
- Boeckhaus, J.; Gross, O. Sodium-Glucose Cotransporter-2 Inhibitors in Patients with Hereditary Podocytopathies, Alport Syndrome, and FSGS: A Case Series to Better Plan a Large-Scale Study. Cells 2021, 10, 1815. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef]
- Boels, M.G.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; van der Vlag, J.; Koster, A.J.; van Zonneveld, A.J.; van Faassen, E.; Gröne, H.-J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef]
- Shippey, E.A.; Wagler, V.D.; Collamer, A.N. Hydroxychloroquine: An old drug with new relevance. Clevel. Clin. J. Med. 2018, 85, 459–467. [Google Scholar] [CrossRef]
- Godwin, J.G.; Ge, X.; Stephan, K.; Jurisch, A.; Tullius, S.G.; Iacomini, J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc. Natl. Acad. Sci. USA 2010, 107, 14339–14344. [Google Scholar] [CrossRef] [PubMed]
- Nozu, K.; Takaoka, Y.; Kai, H.; Takasato, M.; Yabuuchi, K.; Yamamura, T.; Horinouchi, T.; Sakakibara, N.; Ninchoji, T.; Nagano, C.; et al. Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome. Kidney Res. Clin. Pr. 2020, 39, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Rheault, M.N.; Savige, J.; Randles, M.J.; Weinstock, A.; Stepney, M.; Turner, A.N.; Parziale, G.; Gross, O.; A Flinter, F.; Miner, J.H.; et al. The importance of clinician, patient and researcher collaborations in Alport syndrome. Pediatr. Nephrol. 2019, 35, 733–742. [Google Scholar] [CrossRef]
- Arrondel, C.; Deschênes, G.; Le Meur, Y.; Viau, A.; Cordonnier, C.; Fournier, A.; Amadeo, S.; Gubler, M.-C.; Antignac, C.; Heidet, L. A large tandem duplication within the COL4A5 gene is responsible for the high prevalence of Alport syndrome in French Polynesia. Kidney Int. 2004, 65, 2030–2040. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.F.; Hostikka, S.L.; Zhou, J.; Chow, L.T.; Oliphant, A.R.; Gerken, S.C.; Gregory, M.C.; Skolnick, M.H.; Atkin, C.L.; Tryggvason, K. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 1990, 248, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Storey, H.; Cheong, H.I.; Kang, H.G.; Park, E.; Hilbert, P.; Persikov, A.; Torres-Fernandez, C.; Ars, E.; Torra, R.; et al. X-linked and autosomal recessive Alport syndrome: Pathogenic variant features and further genotype-phenotype correlations. PLoS ONE 2016, 11, e0161802. [Google Scholar]
- Inoue, Y.; Nishio, H.; Shirakawa, T.; Nakanishi, K.; Nakamura, H.; Sumino, K.; Nishiyama, K.; Iijima, K.; Yoshikawa, N. Detection of mutations in the COL4A5 gene in over 90% of male patients with X-linked Alport’s syndrome by RT-PCR and direct sequencing. Am. J. Kidney Dis. 1999, 34, 854–862. [Google Scholar] [PubMed]
- Gale, D.P.; Oygar, D.D.; Lin, F.; Oygar, P.D.; Khan, N.; Connor, T.M.; Lapsley, M.; Maxwell, P.H.; Neild, G.H. A novel COL4A1 frameshift mutation in familial kidney disease: The importance of the C-terminal NC1 domain of type IV collagen. Nephrol. Dial. Transplant. 2016, 31, 1908–1914. [Google Scholar] [CrossRef]
- Savige, J.; Huang, M.; Croos Dabrera, M.S.; Shukla, K.; Gibson, J. Genotype-phenotype correlations for pathogenic COL4A3–COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome. Front. Med. 2022, 9, 865034. [Google Scholar] [CrossRef]
- Savige, J.; Ariani, F.; Mari, F.; Bruttini, M.; Renieri, A.; Gross, O.; Deltas, C.; Flinter, F.; Ding, J.; Gale, D.P.; et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr. Nephrol. 2019, 34, 1175–1189. [Google Scholar]
- Savige, J.; Renieri, A.; Ars, E.; Daga, S.; Pinto, A.M.; Rothe, H.; Gale, D.P.; Aksenova, M.; Cerkauskaite, A.; Bielska, O.; et al. Digenic Alport syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 1697–1706. [Google Scholar] [CrossRef]
- Savige, J.; Rana, K.; Tonna, S.; Buzza, M.; Dagher, H.; Wang, Y.Y. Thin basement membrane nephropathy. Kidney Int. 2003, 64, 1169–1178. [Google Scholar] [CrossRef]
- Kamiyoshi, N.; Nozu, K.; Fu, X.J.; Morisada, N.; Nozu, Y.; Ye, M.J.; Imafuku, A.; Miura, K.; Yamamura, T.; Minamikawa, S.; et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 1441. [Google Scholar] [CrossRef]
- Voskarides, K.; Papagregoriou, G.; Hadjipanagi, D.; Petrou, I.; Savva, I.; Elia, A.; Athanasiou, Y.; Pastelli, A.; Kkolou, M.; Hadjigavriel, M.; et al. COL4A5 and LAMA5 variants co-inherited in familial hematuria: Digenic inheritance or genetic modifier effect? BMC Nephrol. 2018, 19, 114. [Google Scholar] [CrossRef]
- Fallerini, C.; Baldassarri, M.; Trevisson, E.; Morbidoni, V.; La Manna, A.; Lazzarin, R.; Pasini, A.; Barbano, G.; Pinciaroli, A.; Garosi, G.; et al. Alport syndrome: Impact of digenic inheritance in patients management. Clin. Genet. 2017, 92, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Lipska-Zietkiewicz, B.S.; Watson, E.; Hertz, J.M.; Deltas, C.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; Cerkauskaite, A.; et al. Guidelines for genetic testing and management of Alport syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Chiereghin, C.; Robusto, M.; Mastrangelo, A.; Castorina, P.; Montini, G.; Giani, M.; Duga, S.; Asselta, R.; Soldà, G. Alport syndrome cold cases: Missing mutations identified by exome sequencing and functional analysis. PLoS ONE 2017, 12, e0178630. [Google Scholar]
- Gibson, J.; Fieldhouse, R.; Chan, M.M.; Sadeghi-Alavijeh, O.; Burnett, L.; Izzi, V.; Persikov, A.V.; Gale, D.P.; Storey, H.; Savige, J.; et al. Prevalence Estimates of Predicted Pathogenic COL4A3–COL4A5 Variants in a Population Sequencing Database and Their Implications for Alport Syndrome. J. Am. Soc. Nephrol. 2021, 32, 2273–2290. [Google Scholar] [CrossRef]
- Al-Amer, O.M.; Mir, R.; Hamadi, A.; Alasseiri, M.I.; Altayar, M.A.; AlZamzami, W.; Moawadh, M.; Alatawi, S.; Niaz, H.A.; Oyouni, A.A.A.; et al. Antiapoptotic Gene Genotype and Allele Variations and the Risk of Lymphoma. Cancers 2023, 15, 1012. [Google Scholar] [CrossRef]
- Alzahrani, O.R.; Mir, R.; Alatwi, H.E.; Hawsawi, Y.M.; Alharbi, A.A.; Alessa, A.H.; Albalawi, E.S.; Elfaki, I.; Alalawi, Y.; Moharam, L.; et al. Potential Impact of PI3K-AKT Signaling Pathway Genes, KLF-14, MDM4, miRNAs 27a, miRNA-196a Genetic Alterations in the Predisposition and Progression of Breast Cancer Patients. Cancers 2023, 15, 1281. [Google Scholar] [CrossRef]
- Rath, S.; Hawsawi, Y.M.; Alzahrani, F.; Khan, M.I. Epigenetic regulation of inflammation: The metabolomics connection. Semin. Cell Dev. Biol. 2022; Online ahead of print. [Google Scholar] [CrossRef]
- Alzahrani, O.R.; Alatwi, H.E.; Alharbi, A.A.; Alessa, A.H.; Al-Amer, O.M.; Alanazi, A.F.R.; Shams, A.M.; Alomari, E.; Naser, A.Y.; Alzahrani, F.A.; et al. Identification and Characterization of Novel Mutations in Chronic Kidney Disease (CKD) and Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Saudi Subjects by Whole-Exome Sequencing. Medicina 2022, 58, 1657. [Google Scholar] [CrossRef] [PubMed]
- Jalalah, S.M. Patterns of primary glomerular diseases among adults in the western region of Saudi Arabia. Saudi J. Kidney Dis. Transplant. 2009, 20, 295–299. [Google Scholar]
- Al-Rasheed, S.A. The impact of renal biopsy in the clinical management of childhood renal disease. Saudi J. Kidney Dis. Transplant. 1997, 8, 11–15. [Google Scholar]
- Chavez, E.; Rodriguez, J.; Drexler, Y.; Fornoni, A. Novel therapies for Alport syndrome. Front. Med. 2022, 9, 848389. [Google Scholar]
- Webb, N.J.A.; Shahinfar, S.; Wells, T.G.; Massaad, R.; Gleim, G.W.; Sisk, C.M.; Lam, C. Losartan and enalapril are comparable in reducing proteinuria in children with Alport syndrome. Pediatr. Nephrol. 2013, 28, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Bomback, A.S.; Klemmer, P.J. The incidence and implications of aldosterone breakthrough. Nat. Clin. Pract. Nephrol. 2007, 3, 486–492. [Google Scholar] [CrossRef]
- Oka, T.; Sakaguchi, Y.; Hattori, K.; Asahina, Y.; Kajimoto, S.; Doi, Y.; Kaimori, J.-Y.; Isaka, Y. Mineralocorticoid receptor antagonist use and hard renal outcomes in real-world patients with chronic kidney disease. Hypertension 2022, 79, 679–689. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Liu, J.; Cui, J.; Fang, X.; Chen, J.; Yan, W.; Shen, Q.; Xu, H. Efficacy and safety of dapagliflozin in children with inherited proteinuric kidney disease: A pilot study. Kidney Int. Rep. 2022, 7, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Strippoli, G.F.; Craig, J.C. KHA-CARI commentary on the KDIGO clinical practice guideline for lipid management in chronic kidney disease. Nephrology 2014, 19, 663–666. [Google Scholar] [CrossRef]
- Esmeijer, K.; Dekkers, O.M.; de Fijter, J.W.; Dekker, F.W.; Hoogeveen, E.K. Effect of different types of statins on kidney function decline and proteinuria: A network meta-analysis. Sci. Rep. 2019, 9, 16632. [Google Scholar]
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68. [Google Scholar]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti–microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Koepke, M.L.; Beirowski, B.; Schulze-Lohoff, E.; Segerer, S.; Weber, M. Nephroprotection by antifibrotic and anti-inflammatory effects of the vasopeptidase inhibitor AVE7688. Kidney Int. 2005, 68, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Bottiglio, C.; Kumar, N.; Maeshima, Y.; Strutz, F.; Müller, G.A.; Kalluri, R.; Takayanagi, K.; Shimizu, T.; Tayama, Y.; et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Ren. Physiol. 2003, 285, F1060–F1067. [Google Scholar] [CrossRef] [PubMed]
- Ninichuk, V.; Gross, O.; Reichel, C.; Khandoga, A.; Pawar, R.D.; Ciubar, R.; Segerer, S.; Belemezova, E.; Radomska, E.; Luckow, B.; et al. Anders HJ. Faculty Opinions recommendation of Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J. Am. Soc. Nephrol. 2005, 16, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Sund, M.; Xie, L.; Cosgrove, D.; Kalluri, R. Faculty Opinions recommendation of Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 7321–7326. [Google Scholar] [CrossRef]
- Hawsawi, Y.M.; Shams, A.; Theyab, A.; Siddiqui, J.; Barnawee, M.; Abdali, W.A.; Marghalani, N.A.; Alshelali, N.H.; Al-Sayed, R.; Alzahrani, O.; et al. The State-of-the-Art of Gene Editing and its Application to Viral Infections and Diseases Including COVID-19. Front. Cell. Infect. Microbiol. 2022, 12, 869889. [Google Scholar]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [PubMed]
- Daga, S.; Donati, F.; Capitani, K.; Croci, S.; Tita, R.; Giliberti, A.; Valentino, F.; Benetti, E.; Fallerini, C.; Niccheri, F.; et al. Correction: New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet. 2020, 28, 480–490, Erratum in Eur. J. Hum. Genet. 2023, Online ahead of print. [Google Scholar] [CrossRef]
- Lin, X.; Suh, J.H.; Go, G.; Miner, J.H. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J. Am. Soc. Nephrol. 2014, 25, 687. [Google Scholar] [CrossRef]
- Funk, S.D.; Bayer, R.H.; Miner, J.H. Endothelial cell-specific collagen type IV-α3 expression does not rescue Alport syndrome in Col4a3−/− mice. Am. J. Physiol. Ren. Physiol. 2019, 316, F830–F837. [Google Scholar] [CrossRef]
- Petzold, F.; Bachmann, A.; Bergmann, C.; Helmchen, U.; Halbritter, J. Retrospective genetic analysis illustrates the spectrum of autosomal Alport syndrome in a case of living-related donor kidney transplantation. BMC Nephrol. 2019, 20, 340. [Google Scholar] [CrossRef]
- Kashtan, C.E. Renal transplantation in patients with Alport syndrome: Patient selection, outcomes, and donor evaluation. Int. J. Nephrol. Renov. Dis. 2018, 11, 267–270. [Google Scholar] [CrossRef] [PubMed]
- El-Hazmi, A.M.; Warsy, A.S.; Al-Swailem, A.M.; Sulaimani, R.; Al-Meshari, A.A. Consanguinity among the Saudi Arabian population. J. Med. Genet. 1995, 32, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Alrefaei, A.F.; Hawsawi, Y.M.; Almaleki, D.; Alafif, T.; Alzahrani, F.A.; Bakhrebah, M.A. Genetic data sharing and artificial intelligence in the era of personalized medicine based on a cross-sectional analysis of the Saudi human genome program. Sci. Rep. 2022, 12, 1–10. [Google Scholar]
- Pajari, H.; Sinkkonen, J. Psychosocial impact of an X-linked hereditary disease: A study of Alport syndrome patients and family members. Child Care Heal. Dev. 2000, 26, 239–250. [Google Scholar] [CrossRef]
- Alsohime, F.; Almaghamsi, T.; Basha, T.A.; Alardati, H.; Alghamdi, M.; Hawsawi, Y.M. Unusual Prominent Pulmonary Involvement in a Homozygous PRF1 Gene Variant in a Female Patient. J. Clin. Immunol. 2020, 41, 217–220. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahrous, N.N.; Jamous, Y.F.; Almatrafi, A.M.; Fallatah, D.I.; Theyab, A.; Alanati, B.H.; Alsagaby, S.A.; Alenazi, M.K.; Khan, M.I.; Hawsawi, Y.M. A Current Landscape on Alport Syndrome Cases: Characterization, Therapy and Management Perspectives. Biomedicines 2023, 11, 2762. https://doi.org/10.3390/biomedicines11102762
Mahrous NN, Jamous YF, Almatrafi AM, Fallatah DI, Theyab A, Alanati BH, Alsagaby SA, Alenazi MK, Khan MI, Hawsawi YM. A Current Landscape on Alport Syndrome Cases: Characterization, Therapy and Management Perspectives. Biomedicines. 2023; 11(10):2762. https://doi.org/10.3390/biomedicines11102762
Chicago/Turabian StyleMahrous, Nahed N., Yahya F. Jamous, Ahmad M. Almatrafi, Deema I. Fallatah, Abdulrahman Theyab, Bayan H. Alanati, Suliman A. Alsagaby, Munifa K. Alenazi, Mohammed I. Khan, and Yousef M. Hawsawi. 2023. "A Current Landscape on Alport Syndrome Cases: Characterization, Therapy and Management Perspectives" Biomedicines 11, no. 10: 2762. https://doi.org/10.3390/biomedicines11102762
APA StyleMahrous, N. N., Jamous, Y. F., Almatrafi, A. M., Fallatah, D. I., Theyab, A., Alanati, B. H., Alsagaby, S. A., Alenazi, M. K., Khan, M. I., & Hawsawi, Y. M. (2023). A Current Landscape on Alport Syndrome Cases: Characterization, Therapy and Management Perspectives. Biomedicines, 11(10), 2762. https://doi.org/10.3390/biomedicines11102762