


Epigenetics in Heart Failure: Role of DNA Methylation in Potential Pathways Leading to Heart Failure with Preserved Ejection Fraction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNA Methylation
3. DNA Methylation in Cardiomyocytes and the Heart
4. Myocardial Fibrosis
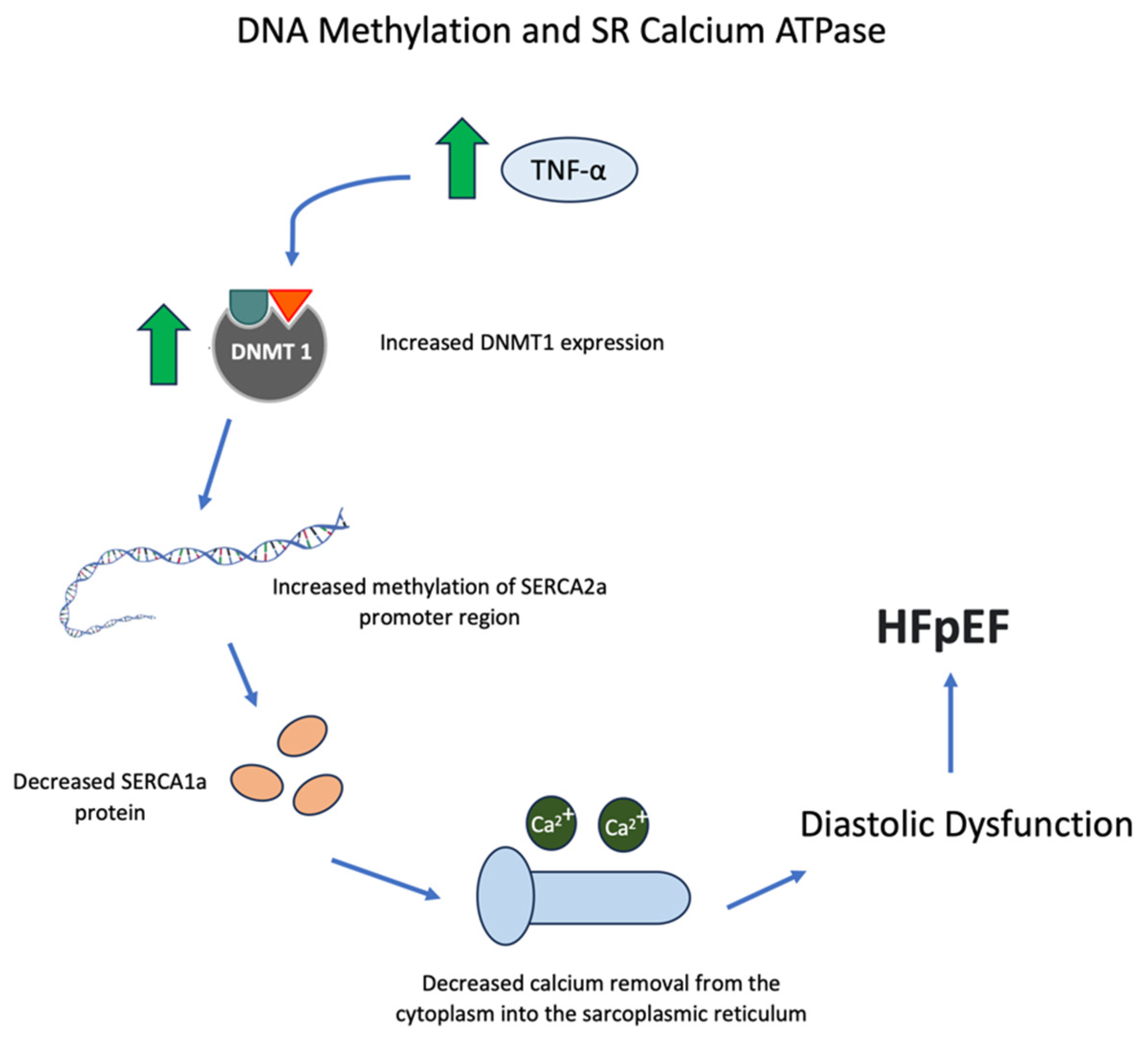
5. SR Calcium ATPase
6. Myocardial Inflammation
7. Mitochondrial and Metabolic Defects
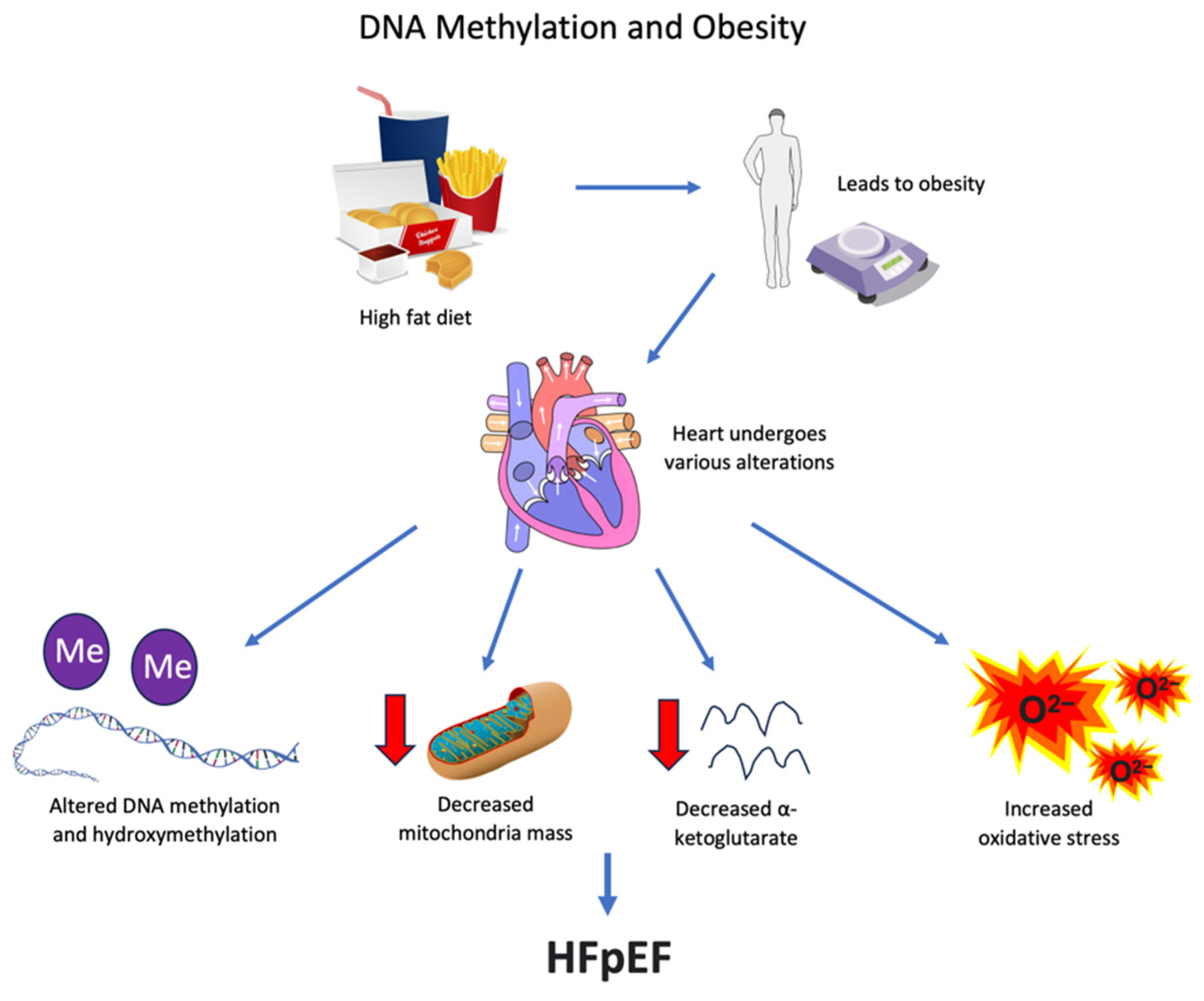
8. Obesity
9. Aging, Atrial Fibrillation, and Other Comorbidities of Patients with HFpEF
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 79, e263–e421. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W. Evaluating the adverse outcome of subtypes of heart failure with preserved ejection fraction defined by machine learning: A systematic review focused on defining high risk phenogroups. EXCLI. J. 2022, 21, 487–518. [Google Scholar]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F. Heart failure with preserved ejection fraction: Insights into diagnosis and pathophysiology. Cardiovasc. Res. 2021, 117, 999–1014. [Google Scholar] [CrossRef]
- Borlaug, B.A.; Paulus, W.J. Heart failure with preserved ejection fraction: Pathophysiology, diagnosis, and treatment. Eur. Heart J. 2011, 32, 670–679. [Google Scholar] [CrossRef]
- Ambrosini, S.; Gorica, E.; Mohammed, S.A.; Costantino, S.; Ruschitzka, F.; Paneni, F. Epigenetic remodeling in heart failure with preserved ejection fraction. Curr. Opin. Cardiol. 2022, 37, 219–226. [Google Scholar] [CrossRef]
- Lv, M.; Ge, W.; Li, Z.; Wang, C.; Zhang, Y. DNA Methylation in Atrial Fibrillation and Its Potential Role in Precision Medicine. Methods Mol. Biol. 2020, 2204, 123–131. [Google Scholar]
- Berezin, A. Epigenetics in heart failure phenotypes. BBA Clin. 2016, 6, 31–37. [Google Scholar] [CrossRef]
- Papait, R.; Greco, C.; Kunderfranco, P.; Latronico, M.V.G.; Condorelli, G. Epigenetics: A new mechanism of regulation of heart failure? Basic Res. Cardiol. 2013, 108, 361. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Costantino, S.; Mügge, A.; Lebeche, D.; Tschöpe, C.; Thum, T.; Paneni, F. Leveraging clinical epigenetics in heart failure with preserved ejection fraction: A call for individualized therapies. Eur. Heart J. 2021, 42, 1940–1958. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sui, Y.; Ruan, X.; Wang, X.; He, K.; Dong, W.; Qu, H.; Fang, X. A deep learning model for early risk prediction of heart failure with preserved ejection fraction by DNA methylation profiles combined with clinical features. Clin. Epigenetics 2022, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Mao, C.; Ding, Y.; Rui, C.; Wu, L.; Shi, A.; Zhang, H.; Zhang, L.; Xu, Z. Molecular and enzymatic profiles of mammalian DNA methyltransferases: Structures and targets for drugs. Curr. Med. Chem. 2010, 17, 4052–4071. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef]
- Hermann, A.; Gowher, H.; Jeltsch, A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004, 61, 2571–2587. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Nührenberg, T.G.; Hammann, N.; Schnick, T.; Preißl, S.; Witten, A.; Stoll, M.; Gilsbach, R.; Neumann, F.-J.; Hein, L. Cardiac Myocyte De Novo DNA Methyltransferases 3a/3b Are Dispensable for Cardiac Function and Remodeling after Chronic Pressure Overload in Mice. PLoS ONE 2015, 10, e0131019. [Google Scholar] [CrossRef]
- Madsen, A.; Höppner, G.; Krause, J.; Hirt, M.N.; Laufer, S.D.; Schweizer, M.; Hansen, A.; Nikolaev, V.O.; Foo, R.S.Y.; Eschenhagen, T.; et al. An Important Role for DNMT3A-Mediated DNA Methylation in Cardiomyocyte Metabolism and Contractility. Circulation 2020, 142, 1562–1578. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and Non-CpG Methylation in Epigenetic Gene Regulation and Brain Function. Genes 2017, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Gruning, B.A.; Kranzhofer, D.; Schneider, P.; Nuhrenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Pepin, M.E.; Drakos, S.; Ha, C.-M.; Tristani-Firouzi, M.; Selzman, C.H.; Fang, J.C.; Wende, A.R.; Wever-Pinzon, O. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H674–H684. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Kennel, P.J.; Liu, B.; Nash, T.R.; Zhuang, R.Z.; Godier-Furnemont, A.F.; Xue, C.; Lu, R.; Colombo, P.C.; Uriel, N.; et al. Effect of mechanical unloading on genome-wide DNA methylation profile of the failing human heart. JCI Insight 2023, 8, e161788. [Google Scholar] [CrossRef]
- Jo, B.-S.; Koh, I.-U.; Bae, J.-B.; Yu, H.-Y.; Jeon, E.-S.; Lee, H.-Y.; Kim, J.-J.; Choi, M.; Choi, S.S. Methylome analysis reveals alterations in DNA methylation in the regulatory regions of left ventricle development genes in human dilated cardiomyopathy. Genomics 2016, 108, 84–92. [Google Scholar] [CrossRef]
- Pepin, M.E.; Ha, C.-M.; Crossman, D.K.; Litovsky, S.H.; Varambally, S.; Barchue, J.P.; Pamboukian, S.V.; Diakos, N.A.; Drakos, S.G.; Pogwizd, S.M.; et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab. Investig. 2019, 99, 371–386. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- Zile, M.R.; Baicu, C.F.; Ikonomidis, J.S.; Stroud, R.E.; Nietert, P.J.; Bradshaw, A.D.; Slater, R.; Palmer, B.M.; Van Buren, P.; Meyer, M.; et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation 2015, 131, 1247–1259. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. Mass Med. Soc. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Tao, H.; Song, Z.-Y.; Ding, X.-S.; Yang, J.-J.; Shi, K.-H.; Li, J. Epigenetic signatures in cardiac fibrosis, special emphasis on DNA methylation and histone modification. Heart Fail. Rev. 2018, 23, 789–799. [Google Scholar] [CrossRef]
- Gyöngyösi, M.; Winkler, J.; Ramos, I.; Do, Q.-T.; Firat, H.; McDonald, K.; González, A.; Thum, T.; Díez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, V.; De Pascale, M.R.; Zullo, A.; Soricelli, A.; Infante, T.; Mancini, F.P.; Napoli, C. Evidence of epigenetic tags in cardiac fibrosis. J. Cardiol. 2017, 69, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Rajgarhia, A.; Ayasolla, K.R.; Zaghloul, N.; Lopez Da Re, J.M.; Miller, E.J.; Ahmed, M. Extracellular Superoxide Dismutase (EC-SOD) Regulates Gene Methylation and Cardiac Fibrosis During Chronic Hypoxic Stress. Front. Cardiovasc Med. 2021, 8, 669975. [Google Scholar] [CrossRef]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef]
- Tao, H.; Yang, J.-J.; Chen, Z.-W.; Xu, S.-S.; Zhou, X.; Zhan, H.-Y.; Shi, K.-H. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology 2014, 323, 42–50. [Google Scholar] [CrossRef]
- Xu, S.-S.; Ding, J.-F.; Shi, P.; Shi, K.-H.; Tao, H. DNMT1-Induced miR-152-3p Suppression Facilitates Cardiac Fibroblast Activation in Cardiac Fibrosis. Cardiovasc. Toxicol. 2021, 21, 984–999. [Google Scholar] [CrossRef]
- Huo, J.-L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.-M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef]
- Inesi, G.; Prasad, A.M.; Pilankatta, R. The Ca2+ ATPase of cardiac sarcoplasmic reticulum: Physiological role and relevance to diseases. Biochem. Biophys. Res. Commun. 2008, 369, 182–187. [Google Scholar] [CrossRef]
- Dally, S.; Corvazier, E.; Bredoux, R.; Bobe, R.; Enouf, J. Multiple and diverse co-expression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J. Mol. Cell. Cardiol. 2010, 48, 633–644. [Google Scholar] [CrossRef]
- Zhihao, L.; Jingyu, N.; Lan, L.; Michael, S.; Rui, G.; Xiyun, B.; Xiaozhi, L.B.; Guanwei, F. SERCA2a: A key protein in the Ca2+ cycle of the heart failure. Heart Fail Rev. 2020, 25, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-K.; Lee, J.-K.; Chiang, F.-T.; Yang, C.-H.; Huang, S.-W.; Hwang, J.-J.; Lin, J.-L.; Tseng, C.-D.; Chen, J.-J.; Tsai, C.-T. Plasma levels of tumor necrosis factor-alpha and interleukin-6 are associated with diastolic heart failure through downregulation of sarcoplasmic reticulum Ca2+ ATPase. Crit. Care Med. 2011, 39, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Zarain-Herzberg, A.; Estrada-Avilés, R.; Fragoso-Medina, J. Regulation of sarco(endo)plasmic reticulum Ca2+-ATPase and calsequestrin gene expression in the heart. Can. J. Physiol. Pharmacol. 2012, 90, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.-H.; Chen, Y.-C.; Cheng, C.-C.; Lee, T.-I.; Chen, Y.-J.; Chen, S.-A. Tumor necrosis factor-alpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit. Care Med. 2010, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef]
- Kao, Y.-H.; Cheng, C.-C.; Chen, Y.-C.; Chung, C.-C.; Lee, T.-I.; Chen, S.-A.; Chen, Y.-J. Hydralazine-induced promoter demethylation enhances sarcoplasmic reticulum Ca2+-ATPase and calcium homeostasis in cardiac myocytes. Lab. Investig. 2011, 91, 1291–1297. [Google Scholar] [CrossRef]
- LaPenna, K.B.; Li, Z.; Doiron, J.E.; Sharp, T.E., 3rd; Xia, H.; Moles, K.; Koul, K.; Wang, J.S.; Polhemus, D.J.; Goodchild, T.T.; et al. Combination Sodium Nitrite and Hydralazine Therapy Attenuates Heart Failure with Preserved Ejection Fraction Severity in a “2-Hit” Murine Model. J. Am. Heart Assoc. 2023, 12, e028480. [Google Scholar] [CrossRef]
- Zamani, P.; Akers, S.; Soto-Calderon, H.; Beraun, M.; Koppula, M.R.; Varakantam, S.; Rawat, D.; Shiva-Kumar, P.; Haines, P.G.; Chittams, J.; et al. Isosorbide Dinitrate, with or Without Hydralazine, Does Not Reduce Wave Reflections, Left Ventricular Hypertrophy, or Myocardial Fibrosis in Patients with Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e004262. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Jeong, H.-Y.; Kang, W.S.; Hong, M.H.; Jeong, H.C.; Shin, M.-G.; Jeong, M.H.; Kim, Y.S.; Ahn, Y. 5-Azacytidine modulates interferon regulatory factor 1 in macrophages to exert a cardioprotective effect. Sci. Rep. 2015, 5, 15768. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell. Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, R.; Takada, S.; Yamashita, Y.; Choi, Y.L.; Nonaka-Sarukawa, M.; Soda, M.; Misawa, Y.; Isomura, T.; Shimada, K.; Mano, H.; et al. Genome-wide histone methylation profile for heart failure. Genes Cells 2009, 14, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W.; Damen, J.E.; Goutsouliak, V.; Krystal, G. Cardiac hypertrophy in the Dahl rat is associated with increased tyrosine phosphorylation of several cytosolic proteins, including a 120 kDa protein. Am. J. Hypertens. 1996, 9, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Yim, J.; Cho, H.; Rabkin, S.W. Gene expression and gene associations during the development of heart failure with preserved ejection fraction in the Dahl salt sensitive model of hypertension. Clin. Exp. Hypertens. 2018, 40, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Minamino, T.; Toko, H.; Sakamoto, M.; Shimizu, I.; Takahashi, H.; Okada, S.; Tateno, K.; Moriya, J.; Yokoyama, M.; et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J. Exp. Med. 2009, 206, 1565–1574. [Google Scholar] [CrossRef]
- Kühn, H.; O’Donnell, V.B. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006, 45, 334–356. [Google Scholar] [CrossRef]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase: Regulation of expression and enzyme activity. Trends Biochem. Sci. 2007, 32, 332–341. [Google Scholar] [CrossRef]
- Dzitoyeva, S.; Imbesi, M.; Ng, L.W.; Manev, H. 5-Lipoxygenase DNA methylation and mRNA content in the brain and heart of young and old mice. Neural. Plast. 2009, 2009, 209596. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, C.; Manev, H. DNA methylation as an epigenetic regulator of neural 5-lipoxygenase expression: Evidence in human NT2 and NT2-N cells. J. Neurochem. 2004, 88, 1424–1430. [Google Scholar] [CrossRef]
- Uhl, J.; Klan, N.; Rose, M.; Entian, K.-D.; Werz, O.; Steinhilber, D. The 5-lipoxygenase promoter is regulated by DNA methylation. J. Biol. Chem. 2002, 277, 4374–4379. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Alcaide, P.; Condorelli, G.; Gillette, T.G.; Heymans, S.; Jones, E.A.V.; Kallikourdis, M.; Lichtman, A.; Marelli-Berg, F.; Shah, S.; et al. Immunometabolic Mechanisms of Heart Failure with Preserved Ejection Fraction. Nat. Cardiovasc. Res. 2022, 1, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure with Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell. Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef]
- Phan, T.; Khalid, A.; Ganesh, N.S.; Gnanadevan, M.; Ibrar, A.; Lynne, W.; Girish, D.; Kiran, P.; Paul, S.; Houman, A.; et al. Heart Failure with Preserved Ejection Fraction Is Characterized by Dynamic Impairment of Active Relaxation and Contraction of the Left Ventricle on Exercise and Associated with Myocardial Energy Deficiency. J. Am. Coll. Cardiol. 2009, 54, 402–409. [Google Scholar] [CrossRef]
- Heid, J.; Cencioni, C.; Ripa, R.; Baumgart, M.; Atlante, S.; Milano, G.; Scopece, A.; Kuenne, C.; Guenther, S.; Azzimato, V.; et al. Age-dependent increase of oxidative stress regulates microRNA-29 family preserving cardiac health. Sci. Rep. 2017, 7, 16839. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Gustafsson, Å.B. Mitochondrial quality control in the myocardium: Cooperation between protein degradation and mitophagy. J. Mol. Cell. Cardiol. 2014, 75, 122–130. [Google Scholar] [CrossRef]
- Madsen, A.; Krause, J.; Hoppner, G.; Hirt, M.N.; Tan, W.L.W.; Lim, I.; Hansen, A.; Nikolaev, V.O.; Foo, R.S.Y.; Eschenhagen, T.; et al. Hypertrophic signaling compensates for contractile and metabolic consequences of DNA methyltransferase 3A loss in human cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 154, 115–123. [Google Scholar] [CrossRef]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef]
- Sheeran, F.L.; Angerosa, J.; Liaw, N.Y.; Cheung, M.M.; Pepe, S. Adaptations in Protein Expression and Regulated Activity of Pyruvate Dehydrogenase Multienzyme Complex in Human Systolic Heart Failure. Oxid. Med. Cell. Longev. 2019, 2019, 4532592. [Google Scholar] [CrossRef] [PubMed]
- Mori, J.; Basu, R.; McLean, B.A.; Das, S.K.; Zhang, L.; Patel, V.B.; Wagg, C.S.; Kassiri, Z.; Lopaschuk, G.D.; Oudit, G.Y. Agonist-induced hypertrophy and diastolic dysfunction are associated with selective reduction in glucose oxidation: A metabolic contribution to heart failure with normal ejection fraction. Circ. Heart Fail. 2012, 5, 493–503. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.I.; Handschin, C. How Epigenetic Modifications Drive the Expression and Mediate the Action of PGC-1alpha in the Regulation of Metabolism. Int. J. Mol. Sci. 2019, 20, 5449. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P.-H.; et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell. Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef]
- Jiang, Y.; Xia, W.; Yang, J.; Zhu, Y.; Chang, H.; Liu, J.; Huo, W.; Xu, B.; Chen, X.; Li, Y.; et al. BPA-induced DNA hypermethylation of the master mitochondrial gene PGC-1α contributes to cardiomyopathy in male rats. Toxicology 2015, 329, 21–31. [Google Scholar] [CrossRef]
- Ling, C.; Poulsen, P.; Carlsson, E.; Ridderstråle, M.; Almgren, P.; Wojtaszewski, J.; Beck-Nielsen, H.; Groop, L.; Vaag, A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J. Clin. Investig. 2004, 114, 1518–1526. [Google Scholar] [CrossRef]
- Warbrick, I.; Rabkin, S.W. Hypoxia-inducible factor 1-alpha (HIF-1α) as a factor mediating the relationship between obesity and heart failure with preserved ejection fraction. Obes. Rev. 2019, 20, 701–712. [Google Scholar] [CrossRef]
- Ong, S.-G.; Lee, W.H.; Theodorou, L.; Kodo, K.; Lim, S.Y.; Shukla, D.H.; Briston, T.; Kiriakidis, S.; Ashcroft, M.; Davidson, S.M.; et al. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc. Res. 2014, 104, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-C.; Liu, T.; Meng, X.; Han, Q.; Zhang, M.; Wang, L.-X. Effect of basic fibroblast growth factor on the myocardial expression of hypoxia-inducible factor-1alpha and vascular endothelial growth factor following acute myocardial infarction. Heart Lung Circ. 2013, 22, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Borlaug, B.A.; Kitzman, D.W.; McCulloch, A.D.; Blaxall, B.C.; Agarwal, R.; Chirinos, J.A.; Collins, S.; Deo, R.C.; Gladwin, M.T.; et al. Research Priorities for Heart Failure with Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020, 141, 1001–1026. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Castelli, S.; Ioannilli, L.; Ciriolo, M.R. High Dietary Fat Intake Affects DNA Methylation/Hydroxymethylation in Mouse Heart: Epigenetic Hints for Obesity-Related Cardiac Dysfunction. Mol. Nutr. Food Res. 2019, 63, e1800970. [Google Scholar] [CrossRef]
- Khanna, D.; Khanna, S.; Khanna, P.; Kahar, P.; Patel, B.M. Obesity: A Chronic Low-Grade Inflammation and Its Markers. Cureus 2022, 14, e22711. [Google Scholar] [CrossRef]
- Rabkin, S.W. Epicardial adipose tissue and reactive oxygen species. Syst. Biol. Free. Radic. Antioxid. 2014, 8, 2054. [Google Scholar]
- Samblas, M.; Milagro, F.I.; Martínez, A. DNA methylation markers in obesity, metabolic syndrome, and weight loss. Epigenetics 2019, 14, 421–444. [Google Scholar] [CrossRef]
- Mahmoud, A.M. An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions. Int. J. Mol. Sci. 2022, 23, 1341. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef]
- Roetker, N.S.; Pankow, J.S.; Bressler, J.; Morrison, A.C.; Boerwinkle, E. Prospective Study of Epigenetic Age Acceleration and Incidence of Cardiovascular Disease Outcomes in the ARIC Study (Atherosclerosis Risk in Communities). Circ. Genom. Precis. Med. 2018, 11, e001937. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.B.; Occean, J.R.; Sen, P. Epigenetic dysregulation in cardiovascular aging and disease. J. Cardiovasc. Aging 2021, 1, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Atrial Fibrillation and Heart Failure with Preserved Ejection Fraction in Patients with Nonalcoholic Fatty Liver Disease. Am. J. Med. 2020, 133, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W.; Nourai, H. Atrial fibrillation in heart failure with preserved ejection fraction. J. Atr. Fibrillation 2022, 2, 92–98. [Google Scholar]
- Kao, Y.-H.; Chen, Y.-C.; Chung, C.-C.; Lien, G.-S.; Chen, S.-A.; Kuo, C.-C.; Chen, Y.-J. Heart failure and angiotensin II modulate atrial Pitx2c promotor methylation. Clin. Exp. Pharmacol. Physiol. 2013, 40, 379–384. [Google Scholar] [CrossRef]
- Reilly, L.; Eckhardt, L.L. Cardiac potassium inward rectifier Kir2: Review of structure, regulation, pharmacology, and arrhythmogenesis. Heart. Rhythm. 2021, 18, 1423–1434. [Google Scholar] [CrossRef]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Jin, J.; Zhu, C.; Wang, J.; Zhao, X.; Yang, R. The association between ACTB methylation in peripheral blood and coronary heart disease in a case-control study. Front. Cardiovasc. Med. 2022, 9, 972566. [Google Scholar] [CrossRef]
- Cheng, Y.; Gadd, D.A.; Gieger, C.; Monterrubio-Gómez, K.; Zhang, Y.; Berta, I.; Stam, M.J.; Szlachetka, N.; Lobzaev, E.; Wrobel, N.; et al. Development and validation of DNA methylation scores in two European cohorts augment 10-year risk prediction of type 2 diabetes. Nat. Aging 2023, 3, 450–458. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabkin, S.W.; Wong, C.N. Epigenetics in Heart Failure: Role of DNA Methylation in Potential Pathways Leading to Heart Failure with Preserved Ejection Fraction. Biomedicines 2023, 11, 2815. https://doi.org/10.3390/biomedicines11102815
Rabkin SW, Wong CN. Epigenetics in Heart Failure: Role of DNA Methylation in Potential Pathways Leading to Heart Failure with Preserved Ejection Fraction. Biomedicines. 2023; 11(10):2815. https://doi.org/10.3390/biomedicines11102815
Chicago/Turabian StyleRabkin, Simon W., and Chenille N. Wong. 2023. "Epigenetics in Heart Failure: Role of DNA Methylation in Potential Pathways Leading to Heart Failure with Preserved Ejection Fraction" Biomedicines 11, no. 10: 2815. https://doi.org/10.3390/biomedicines11102815
APA StyleRabkin, S. W., & Wong, C. N. (2023). Epigenetics in Heart Failure: Role of DNA Methylation in Potential Pathways Leading to Heart Failure with Preserved Ejection Fraction. Biomedicines, 11(10), 2815. https://doi.org/10.3390/biomedicines11102815