
An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
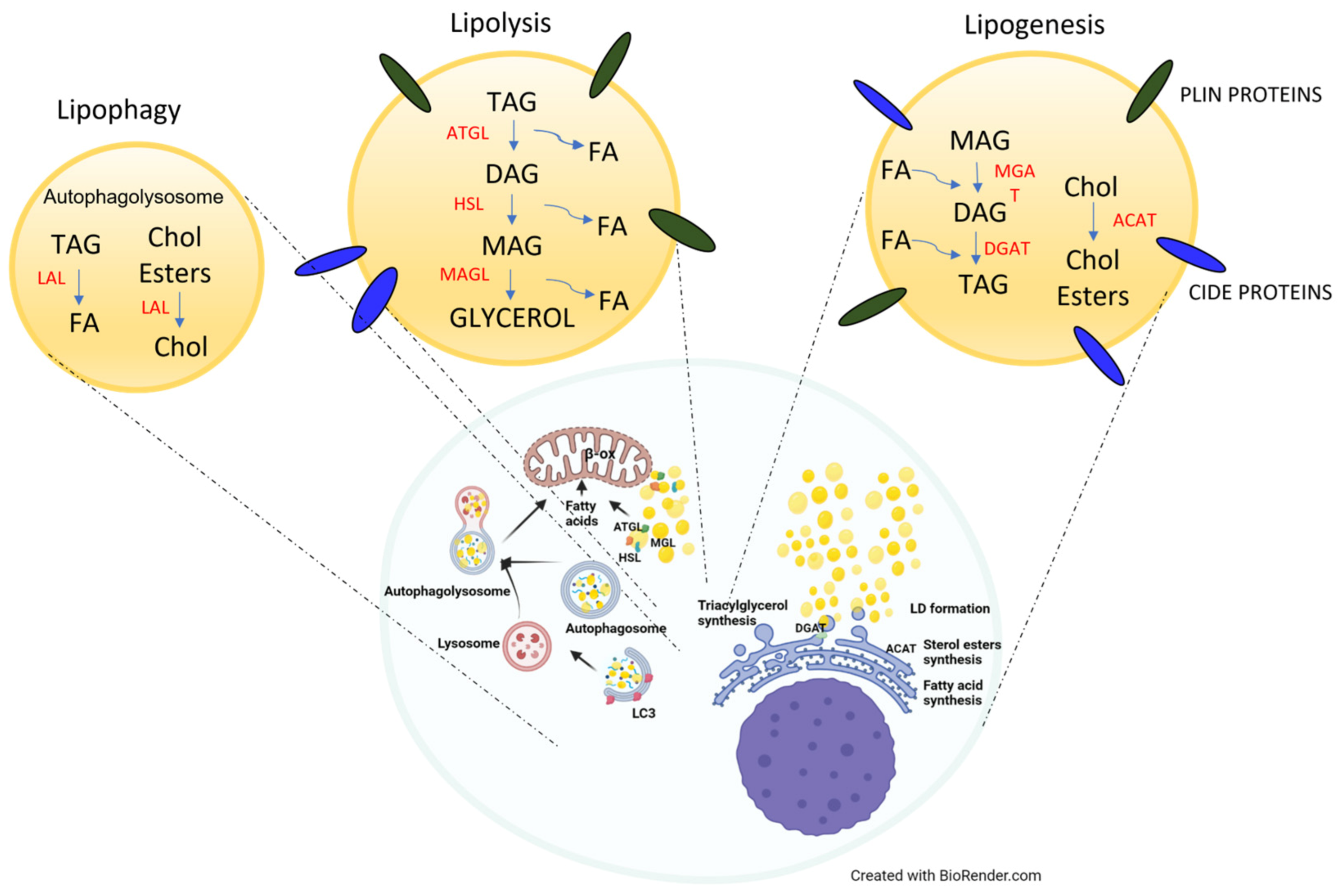
2. Lipid Droplets in Healthy Cells and Cancer Cells
2.1. Physiological Roles of LDs
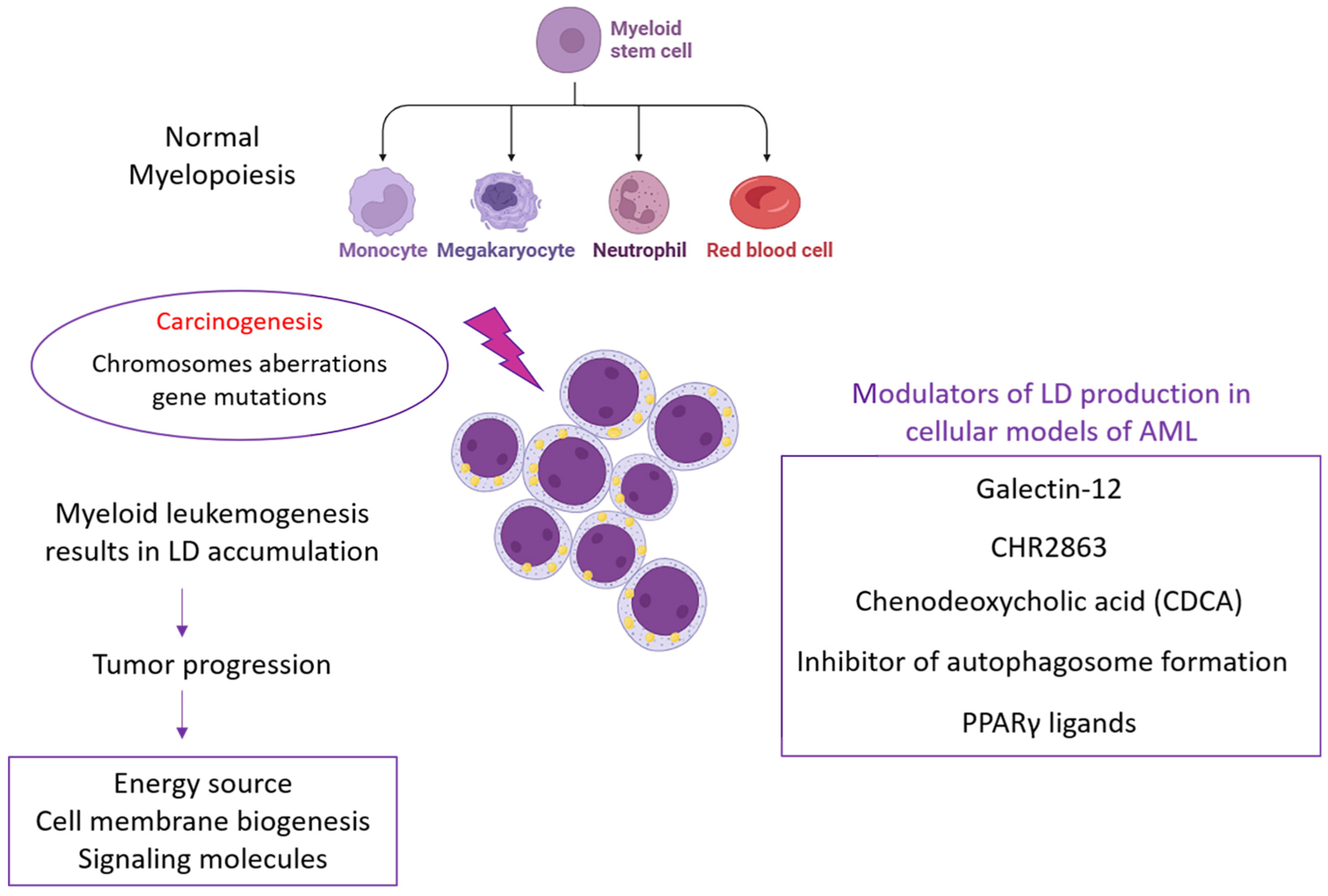
2.2. Pathological Roles of LDs
3. Targeting Lipid Droplets in Acute Myeloid Leukemia
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 698023. [Google Scholar] [CrossRef]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef]
- Tirinato, L.; Pagliari, F.; Limongi, T.; Marini, M.; Falqui, A.; Seco, J.; Candeloro, P.; Liberale, C.; Di Fabrizio, E. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells Int. 2017, 2017, 1656053. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Wu, J.; Ren, Z. Lipid droplets: A cellular organelle vital in cancer cells. Cell Death Discov. 2023, 9, 254. [Google Scholar] [CrossRef]
- Nisticò, C.; Pagliari, F.; Chiarella, E.; Fernandes Guerreiro, J.; Marafioti, M.G.; Aversa, I.; Genard, G.; Hanley, R.; Garcia-Calderón, D.; Bond, H.M.; et al. Lipid Droplet Biosynthesis Impairment through DGAT2 Inhibition Sensitizes MCF7 Breast Cancer Cells to Radiation. Int. J. Mol. Sci. 2021, 22, 10102. [Google Scholar] [CrossRef]
- Tirinato, L.; Marafioti, M.G.; Pagliari, F.; Jansen, J.; Aversa, I.; Hanley, R.; Nisticò, C.; Garcia-Calderón, D.; Genard, G.; Guerreiro, J.F.; et al. Lipid droplets and ferritin heavy chain: A devilish liaison in human cancer cell radioresistance. eLife 2021, 10, e72943. [Google Scholar] [CrossRef]
- Li, D.; Liang, J.; Yang, W.; Guo, W.; Song, W.; Zhang, W.; Wu, X.; He, B. A distinct lipid metabolism signature of acute myeloid leukemia with prognostic value. Front. Oncol. 2022, 12, 876981. [Google Scholar] [CrossRef]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862 Pt B, 1260–1272. [Google Scholar] [CrossRef]
- Apffel, C.A.; Baker, J.R. Lipid droplets in the cytoplasm of malignant cells. Cancer 1964, 17, 176–184. [Google Scholar] [CrossRef]
- Novikoff, A.B. A transplantable rat liver tumor induced by 4-dimethylaminoazobenzene. Cancer Res. 1957, 17, 1010–1027. [Google Scholar]
- Antunes, P.; Cruz, A.; Barbosa, J.; Bonifácio, V.D.B.; Pinto, S.N. Lipid Droplets in Cancer: From Composition and Role to Imaging and Therapeutics. Molecules 2022, 27, 991. [Google Scholar] [CrossRef]
- Mesuraca, M.; Nisticò, C.; Lombardo, N.; Piazzetta, G.L.; Lobello, N.; Chiarella, E. Cellular and Biochemical Characterization of Mesenchymal Stem Cells from Killian Nasal Polyp. Int. J. Mol. Sci. 2022, 23, 13214. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef]
- Morikawa, K.; Hanada, H.; Hirota, K.; Nonaka, M.; Ikeda, C. All-trans retinoic acid displays multiple effects on the growth, lipogenesis and adipokine gene expression of AML-I preadipocyte cell line. Cell Biol. Int. 2013, 37, 36–46. [Google Scholar] [CrossRef]
- Bilous, N.; Abramenko, I.; Chumak, A.; Dyagil, I.; Martina, Z. Analysis of LPL gene expression in patients with chronic lymphocytic leukemia. Exp. Oncol. 2019, 41, 39–45. [Google Scholar] [CrossRef]
- McKillop, I.H.; Girardi, C.A.; Thompson, K.J. Role of fatty acid binding proteins (FABPs) in cancer development and progression. Cell. Signal. 2019, 62, 109336. [Google Scholar] [CrossRef]
- Sabatier, M.; Birsen, R.; Lauture, L.; Mouche, S.; Angelino, P.; Dehairs, J.; Goupille, L.; Boussaid, I.; Heiblig, M.; Boet, E.; et al. C/EBPα Confers Dependence to Fatty Acid Anabolic Pathways and Vulnerability to Lipid Oxidative Stress-Induced Ferroptosis in FLT3-Mutant Leukemia. Cancer Discov. 2023, 13, 1720–1747. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Esmaeili, S.; Salari, S.; Kaveh, V.; Ghaffari, S.H.; Bashash, D. Alteration of PPAR-GAMMA (PPARG; PPARγ) and PTEN gene expression in acute myeloid leukemia patients and the promising anticancer effects of PPARγ stimulation using pioglitazone on AML cells. Mol. Genet. Genom. Med. 2021, 9, e1818. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef]
- Poirier, S.; Samami, S.; Mamarbachi, M.; Demers, A.; Chang, T.Y.; Vance, D.E.; Hatch, G.M.; Mayer, G. The epigenetic drug 5-azacytidine interferes with cholesterol and lipid metabolism. J. Biol. Chem. 2014, 289, 18736–18751. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Fitchev, P.; Brooks, K.M.; Franco, O.E.; Cheng, K.; Hayward, S.W.; Welte, M.A.; Crawford, S.E. Lipid droplet velocity is a microenvironmental sensor of aggressive tumors regulated by V-ATPase and PEDF. Lab. Investig. 2019, 99, 1822–1834. [Google Scholar] [CrossRef]
- Wang, W.; Bai, L.; Li, W.; Cui, J. The Lipid Metabolic Landscape of Cancers and New Therapeutic Perspectives. Front. Oncol. 2020, 10, 605154. [Google Scholar] [CrossRef] [PubMed]
- Kiyoki, Y.; Kato, T.; Kito, S.; Matsuzaka, T.; Morioka, S.; Sasaki, J.; Makishima, K.; Sakamoto, T.; Nishikii, H.; Obara, N.; et al. The fatty acid elongase Elovl6 is crucial for hematopoietic stem cell engraftment and leukemia propagation. Leukemia 2023, 37, 910–913. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Al, M.; Assaraf, Y.G.; Kammerer, S.; Chandrupatla, D.M.; Honeywell, R.; Musters, R.P.; Giovannetti, E.; O’Toole, T.; Scheffer, G.L.; et al. Multifactorial resistance to aminopeptidase inhibitor prodrug CHR2863 in myeloid leukemia cells: Down-regulation of carboxylesterase 1, drug sequestration in lipid droplets and pro-survival activation ERK/Akt/mTOR. Oncotarget 2016, 7, 5240–5257. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Miao, H.; Cheng, X.L.; Wei, F. Lipidomics: Novel insight into the biochemical mechanism of lipid metabolism and dysregulation-associated disease. Chem. Biol. Interact. 2015, 240, 220–238. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.; Xing, Y.; Si, D.; Wang, S.; Lin, J.; Li, C.; Zhang, J.; Yin, D. Combined metabolomic and lipidomic analysis uncovers metabolic profile and biomarkers for papillary thyroid carcinoma. Sci. Rep. 2023, 13, 17666. [Google Scholar] [CrossRef]
- Bernaudo, F.; Monteleone, F.; Mesuraca, M.; Krishnan, S.; Chiarella, E.; Scicchitano, S.; Cuda, G.; Morrone, G.; Bond, H.M.; Gaspari, M. Validation of a novel shotgun proteomic workflow for the discovery of protein-protein interactions: Focus on ZNF521. J. Proteome Res. 2015, 14, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Petan, T. Lipid Droplets in Cancer. Rev. Physiol. Biochem. Pharmacol. 2023, 185, 53–86. [Google Scholar] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Chung, J.; Farese, R.V. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Beller, M. The why, when and how of lipid droplet diversity. J. Cell Sci. 2017, 130, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Cotte, A.K.; Connat, J.L.; Hermetet, F.; Bouyer, F.; Aires, V. Emergence of Lipid Droplets in the Mechanisms of Carcinogenesis and Therapeutic Responses. Cancers 2023, 15, 4100. [Google Scholar] [CrossRef]
- Pagliari, F.; Marafioti, M.G.; Genard, G.; Candeloro, P.; Viglietto, G.; Seco, J.; Tirinato, L. ssRNA Virus and Host Lipid Rearrangements: Is There a Role for Lipid Droplets in SARS-CoV-2 Infection? Front. Mol. Biosci. 2020, 7, 578964. [Google Scholar] [CrossRef]
- Kassan, A.; Herms, A.; Fernández-Vidal, A.; Bosch, M.; Schieber, N.L.; Reddy, B.J.; Fajardo, A.; Gelabert-Baldrich, M.; Tebar, F.; Enrich, C.; et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 2013, 203, 985–1001. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Olzmann, J.A. Establishing the lipid droplet proteome: Mechanisms of lipid droplet protein targeting and degradation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862 Pt B, 1166–1177. [Google Scholar] [CrossRef]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 97–112.e7. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Shen, W.J.; Patel, S.; Greenberg, A.S.; Azhar, S.; Kraemer, F.B. Fat-specific protein 27 modulates nuclear factor of activated T cells 5 and the cellular response to stress. J. Lipid Res. 2013, 54, 734–743. [Google Scholar] [CrossRef]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shinoda, A.; Furuya, N.; Harada, E.; Arimura, N.; Ichi, I.; Fujiwara, Y.; Inoue, J.; Sato, R. Perilipin-mediated lipid droplet formation in adipocytes promotes sterol regulatory element-binding protein-1 processing and triacylglyceride accumulation. PLoS ONE 2013, 8, e64605. [Google Scholar] [CrossRef]
- Guo, Y.; Cordes, K.R.; Farese, R.V.; Walther, T.C. Lipid droplets at a glance. J. Cell Sci. 2009, 122 Pt 6, 749–752. [Google Scholar] [CrossRef]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef]
- Wilfling, F.; Thiam, A.R.; Olarte, M.J.; Wang, J.; Beck, R.; Gould, T.J.; Allgeyer, E.S.; Pincet, F.; Bewersdorf, J.; Farese, R.V., Jr.; et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. eLife 2014, 3, e01607. [Google Scholar] [CrossRef] [PubMed]
- Chorlay, A.; Monticelli, L.; Veríssimo Ferreira, J.; Ben M’barek, K.; Ajjaji, D.; Wang, S.; Johnson, E.; Beck, R.; Omrane, M.; Beller, M.; et al. Membrane Asymmetry Imposes Directionality on Lipid Droplet Emergence from the ER. Dev. Cell 2019, 50, 25–42.e7. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Rothenberg, A.; Gomez, C.; Cohen, A.W.; Garcia, A.; Bhattacharyya, S.; Shapiro, L.; Dolios, G.; Wang, R.; Lisanti, M.P.; et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 2004, 279, 42062–42071. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Xu, G.; Dorward, H.; Tansey, J.T.; Contreras, J.A.; Kimmel, A.R.; Londos, C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 2003, 161, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS-lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, K.; Sarkadi-Nagy, E.; Duncan, R.E.; Ahmadian, M.; Sul, H.S. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1–G4. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. Lysosome: Regulator of lipid degradation pathways. Trends Cell Biol. 2014, 24, 743–750. [Google Scholar] [CrossRef]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef]
- Farge, T.; Nakhle, J.; Lagarde, D.; Cognet, G.; Polley, N.; Castellano, R.; Nicolau, M.L.; Bosc, C.; Sabatier, M.; Sahal, A.; et al. CD36 Drives Metastasis and Relapse in Acute Myeloid Leukemia. Cancer Res. 2023, 83, 2824–2838. [Google Scholar] [CrossRef]
- Fujimoto, T.; Kogo, H.; Ishiguro, K.; Tauchi, K.; Nomura, R. Caveolin-2 is targeted to lipid droplets, a new “membrane domain” in the cell. J. Cell Biol. 2001, 152, 1079–1085. [Google Scholar] [CrossRef]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Cassara, J.; Weller, P.F. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid-derived cells. Blood 2000, 95, 1078–1085. [Google Scholar] [CrossRef]
- Bozza, P.T.; Viola, J.P. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef]
- Vakiti, A.M.P.; Wood, S.K. Acute Myeloid Leukemia; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chiarella, E.; Aloisio, A.; Scicchitano, S.; Todoerti, K.; Cosentino, E.G.; Lico, D.; Neri, A.; Amodio, N.; Bond, H.M.; Mesuraca, M. ZNF521 Enhances MLL-AF9-Dependent Hematopoietic Stem Cell Transformation in Acute Myeloid Leukemias by Altering the Gene Expression Landscape. Int. J. Mol. Sci. 2021, 22, 10814. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, E.; Nisticò, C.; Di Vito, A.; Morrone, H.L.; Mesuraca, M. Targeting of Mevalonate-Isoprenoid Pathway in Acute Myeloid Leukemia Cells by Bisphosphonate Drugs. Biomedicines 2022, 10, 1146. [Google Scholar] [CrossRef] [PubMed]
- Mesuraca, M.; Nisticò, C.; Chiarella, E. Editorial to the Special Issue “Recent Advances in Biochemical Mechanisms of Acute Myeloid Leukemia”. Biomedicines 2023, 11, 1339. [Google Scholar] [CrossRef] [PubMed]
- Zee, B.M.; Poels, K.E.; Yao, C.H.; Kawabata, K.C.; Wu, G.; Duy, C.; Jacobus, W.D.; Senior, E.; Endress, J.E.; Jambhekar, A.; et al. Combined epigenetic and metabolic treatments overcome differentiation blockade in acute myeloid leukemia. iScience 2021, 24, 102651. [Google Scholar] [CrossRef]
- Logan, C.; Koura, D.; Taplitz, R. Updates in infection risk and management in acute leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 135–139. [Google Scholar] [CrossRef]
- Huber, S.; Baer, C.; Hutter, S.; Dicker, F.; Meggendorfer, M.; Pohlkamp, C.; Kern, W.; Haferlach, T.; Haferlach, C.; Hoermann, G. AML classification in the year 2023: How to avoid a Babylonian confusion of languages. Leukemia 2023, 37, 1413–1420. [Google Scholar] [CrossRef]
- Ha, M.; Han, M.E.; Kim, J.Y.; Jeong, D.C.; Oh, S.O.; Kim, Y.H. Prognostic role of TPD52 in acute myeloid leukemia: A retrospective multicohort analysis. J. Cell. Biochem. 2019, 120, 3672–3678. [Google Scholar] [CrossRef] [PubMed]
- Delaidelli, A.; Dunham, C.; Santi, M.; Negri, G.L.; Triscott, J.; Zheludkova, O.; Golanov, A.; Ryzhova, M.; Okonechnikov, K.; Schrimpf, D.; et al. Clinically Tractable Outcome Prediction of Non-WNT/Non-SHH Medulloblastoma Based on TPD52 IHC in a Multicohort Study. Clin. Cancer Res. 2022, 28, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Zhong, A.; Chen, T.; Zhou, T.; Zhang, Z.; Shi, M. TPD52L2 Is a Prognostic Biomarker and Correlated With Immune Infiltration in Lung Adenocarcinoma. Front. Pharmacol. 2021, 12, 728420. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; DiNardo, C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncol. 2018, 14, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar] [CrossRef] [PubMed]
- Torii, I.; Morikawa, S.; Nakano, A.; Morikawa, K. Establishment of a human preadipose cell line, HPB-AML-I: Refractory to PPARgamma-mediated adipogenic stimulation. J. Cell. Physiol. 2003, 197, 42–52. [Google Scholar] [CrossRef]
- Yang, R.Y.; Hsu, D.K.; Yu, L.; Chen, H.Y.; Liu, F.T. Galectin-12 is required for adipogenic signaling and adipocyte differentiation. J. Biol. Chem. 2004, 279, 29761–29766. [Google Scholar] [CrossRef]
- Xue, H.; Yang, R.Y.; Tai, G.; Liu, F.T. Galectin-12 inhibits granulocytic differentiation of human NB4 promyelocytic leukemia cells while promoting lipogenesis. J. Leukoc. Biol. 2016, 100, 657–664. [Google Scholar] [CrossRef]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef]
- Kökény, G.; Calvier, L.; Hansmann, G. PPARγ and TGFβ-Major Regulators of Metabolism, Inflammation, and Fibrosis in the Lungs and Kidneys. Int. J. Mol. Sci. 2021, 22, 10431. [Google Scholar] [CrossRef]
- Chiarella, E.; Lombardo, N.; Lobello, N.; Piazzetta, G.L.; Morrone, H.L.; Mesuraca, M.; Bond, H.M. Deficit in Adipose Differentiation in Mesenchymal Stem Cells Derived from Chronic Rhinosinusitis Nasal Polyps Compared to Nasal Mucosal Tissue. Int. J. Mol. Sci. 2020, 21, 9214. [Google Scholar] [CrossRef] [PubMed]
- Yasugi, E.; Horiuchi, A.; Uemura, I.; Okuma, E.; Nakatsu, M.; Saeki, K.; Kamisaka, Y.; Kagechika, H.; Yasuda, K.; Yuo, A. Peroxisome proliferator-activated receptor gamma ligands stimulate myeloid differentiation and lipogenensis in human leukemia NB4 cells. Dev. Growth Differ. 2006, 48, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, Y.; Jia, W.; Can, C.; Wang, R.; Yang, X.; Gu, C.; Liu, F.; Ji, C.; Ma, D. Chenodeoxycholic acid suppresses AML progression through promoting lipid peroxidation via ROS/p38 MAPK/DGAT1 pathway and inhibiting M2 macrophage polarization. Redox Biol. 2022, 56, 102452. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nisticò, C.; Chiarella, E. An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy. Biomedicines 2023, 11, 3186. https://doi.org/10.3390/biomedicines11123186
Nisticò C, Chiarella E. An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy. Biomedicines. 2023; 11(12):3186. https://doi.org/10.3390/biomedicines11123186
Chicago/Turabian StyleNisticò, Clelia, and Emanuela Chiarella. 2023. "An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy" Biomedicines 11, no. 12: 3186. https://doi.org/10.3390/biomedicines11123186
APA StyleNisticò, C., & Chiarella, E. (2023). An Overview on Lipid Droplets Accumulation as Novel Target for Acute Myeloid Leukemia Therapy. Biomedicines, 11(12), 3186. https://doi.org/10.3390/biomedicines11123186