
Rhamnetin Prevents Bradykinin-Induced Expression of Matrix Metalloproteinase-9 in Rat Brain Astrocytes by Suppressing Protein Kinase-Dependent AP-1 Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Western Blot
2.4. Gelatin Zymography
2.5. Real-Time PCR Analysis
2.6. Transient siRNA Transfection
2.7. Promoter Assay
2.8. Cell Migration Assay
2.9. Statistical Analysis
3. Results
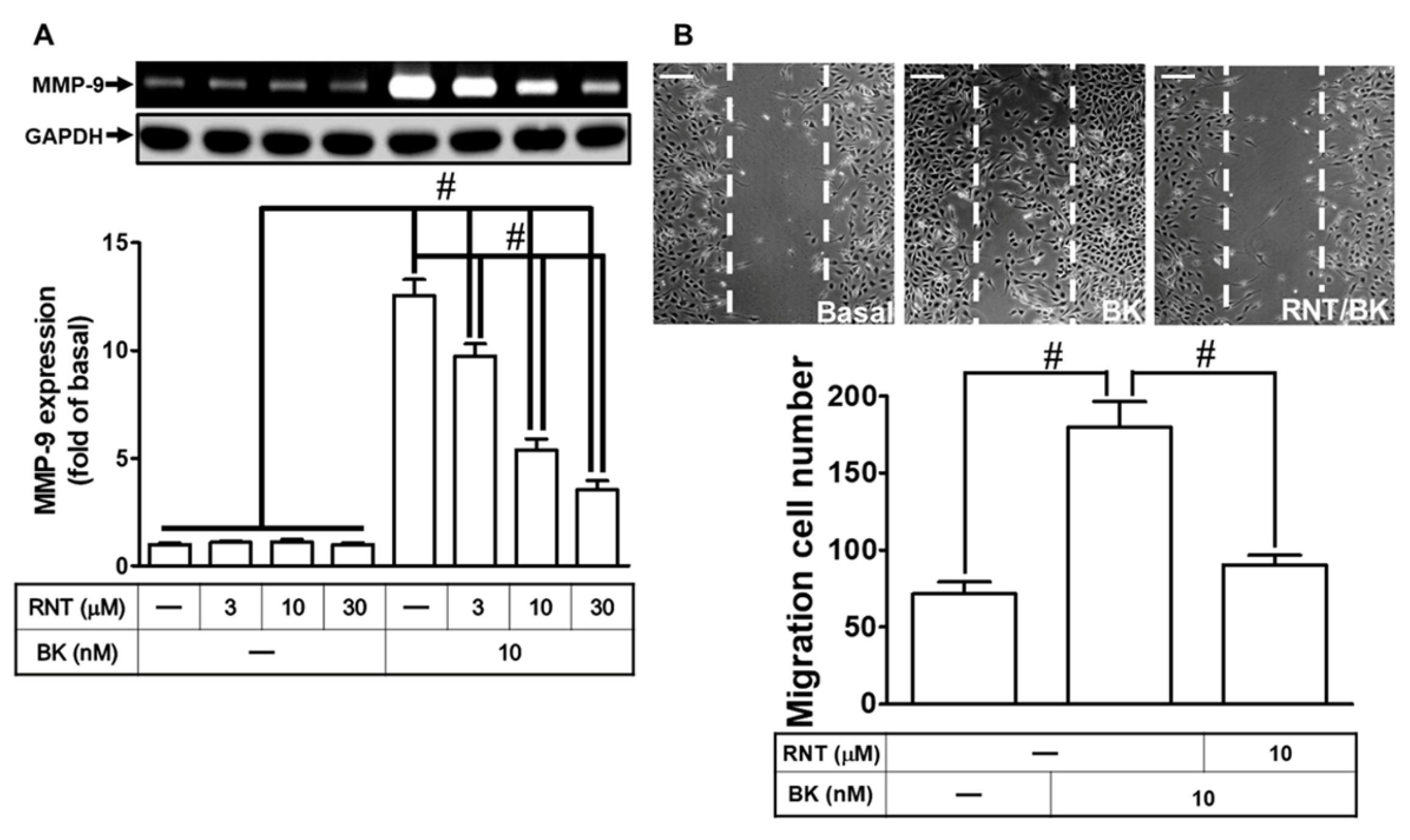
3.1. RNT Suppresses BK-Induced Migration and MMP-9 Up-Regulation
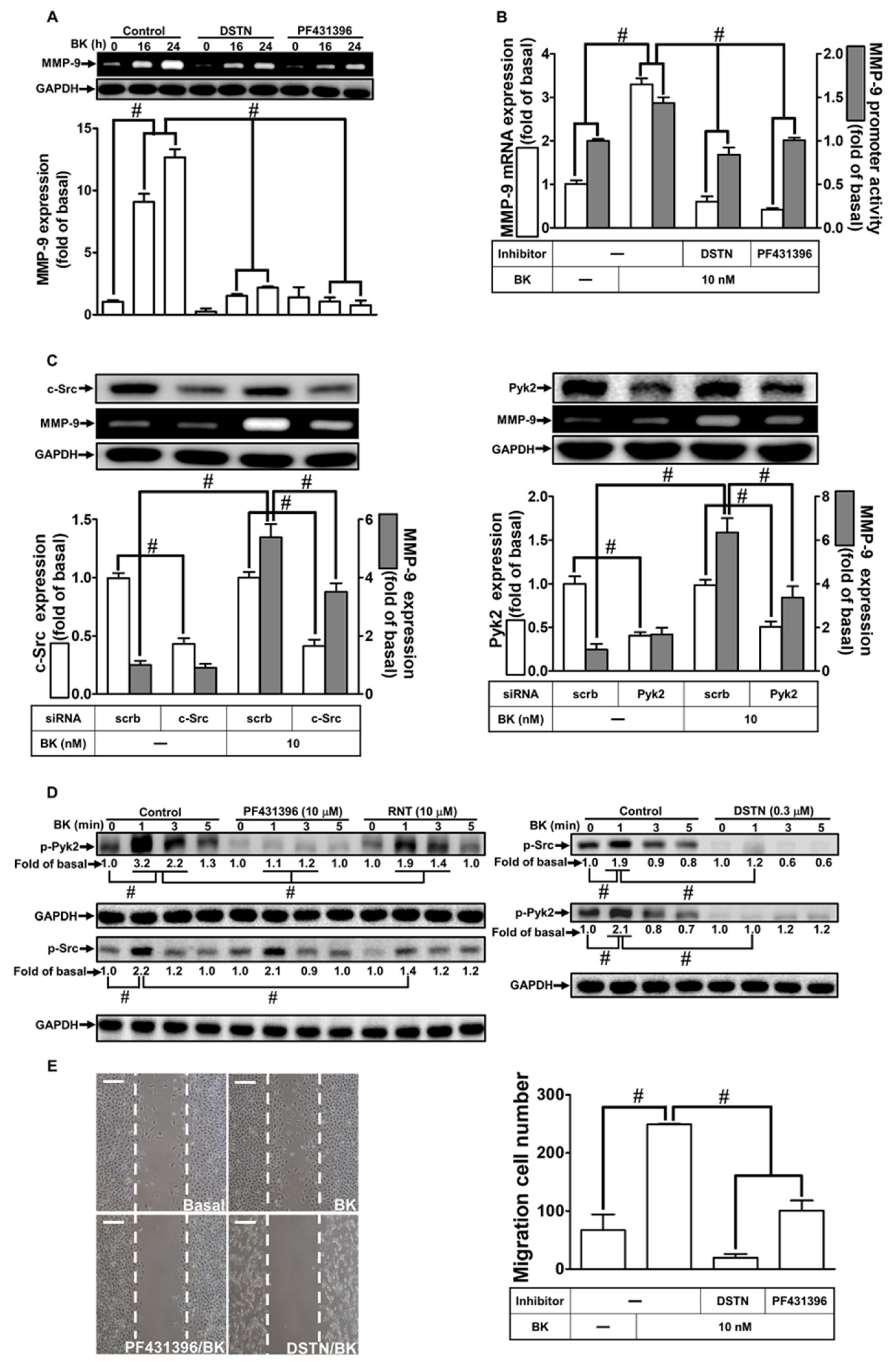
3.2. RNT Suppresses BK-Induced MMP-9 Expression by Inhibiting Pyk2 and c-Src Activation
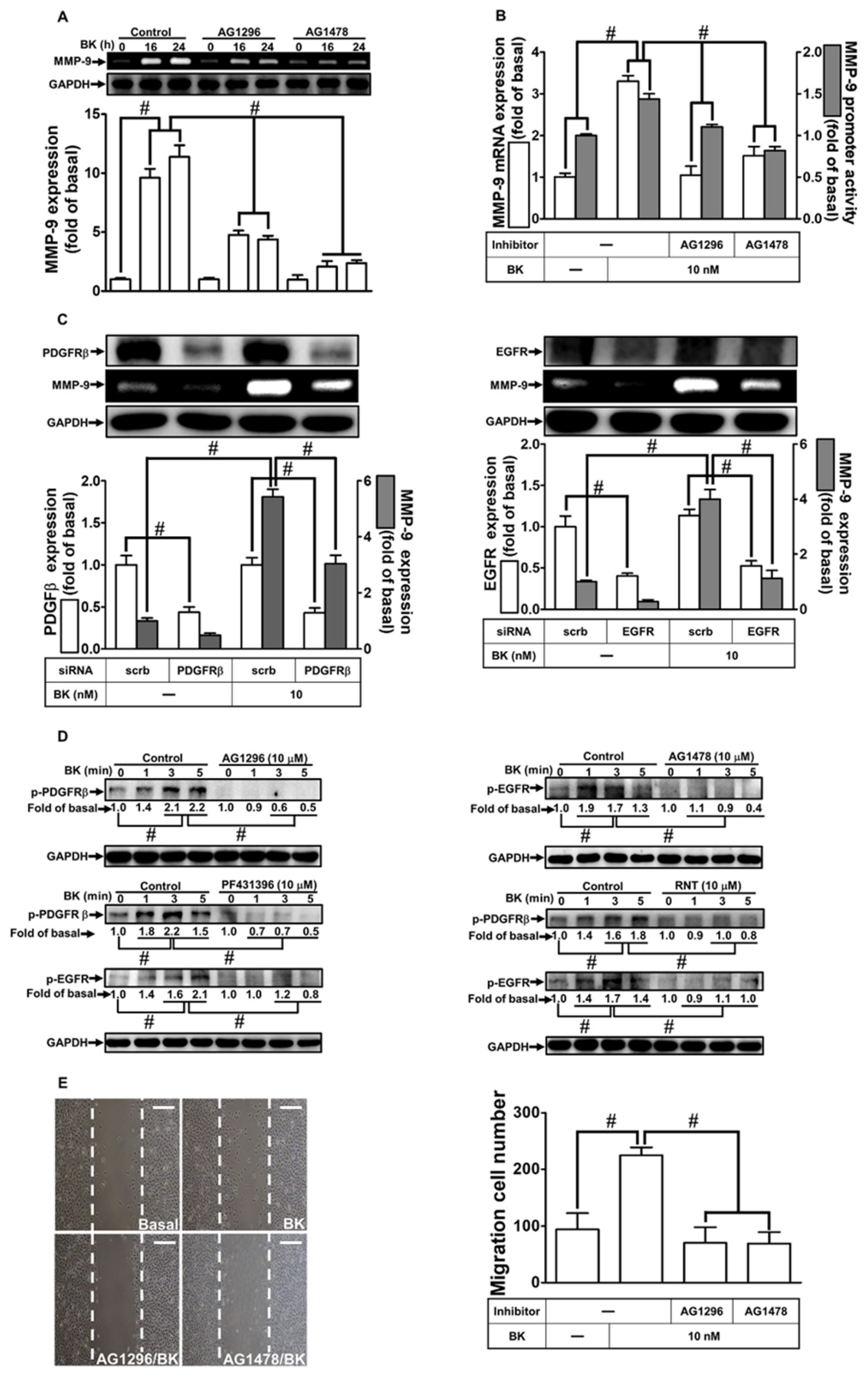
3.3. RNT Suppresses BK-Induced MMP-9 Expression via Inhibition of PDGFR and EGFR Transactivation
3.4. RNT Suppresses BK-Induced MMP-9 Expression and Cell Migration via Inhibition of the PI3K/Akt Pathway
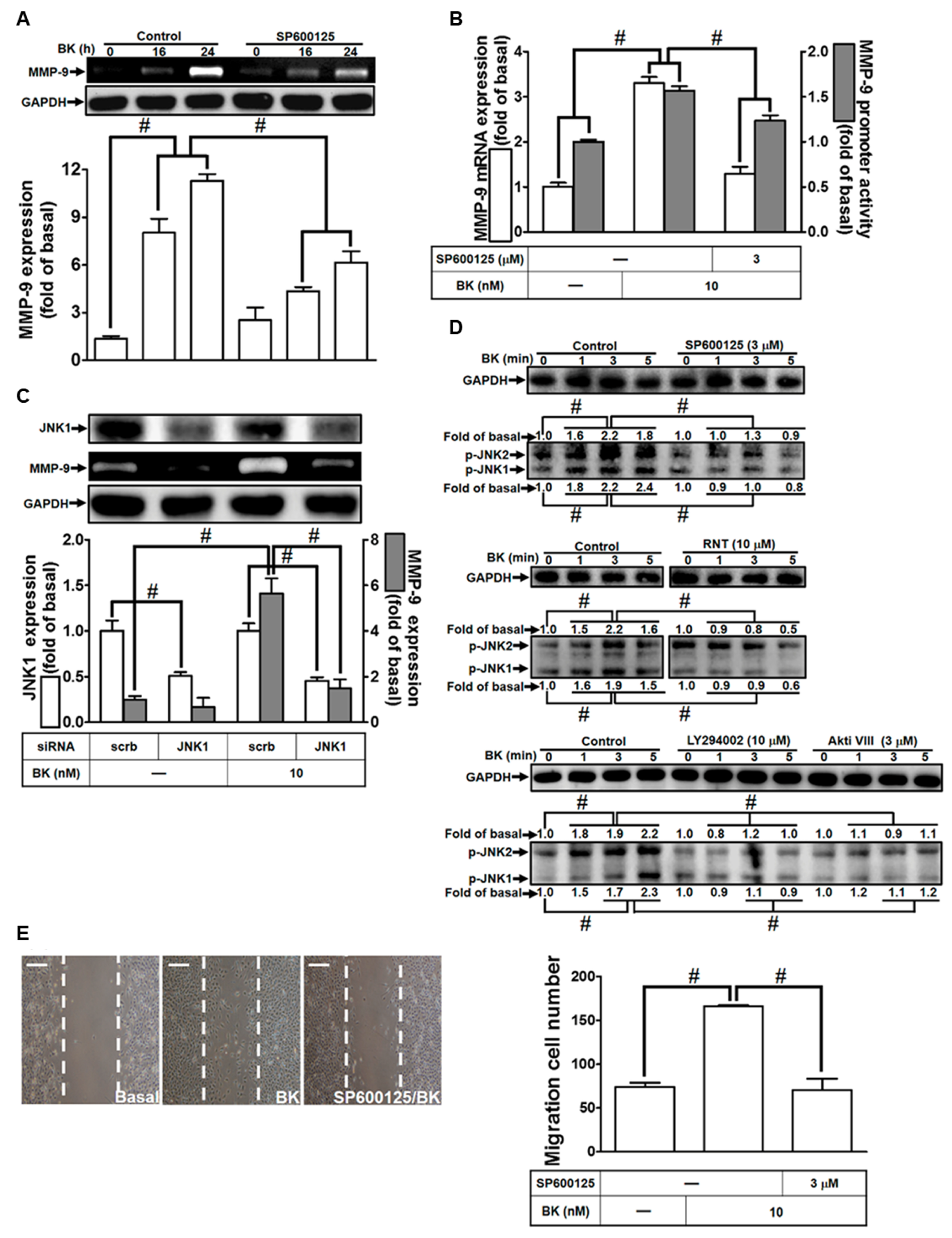
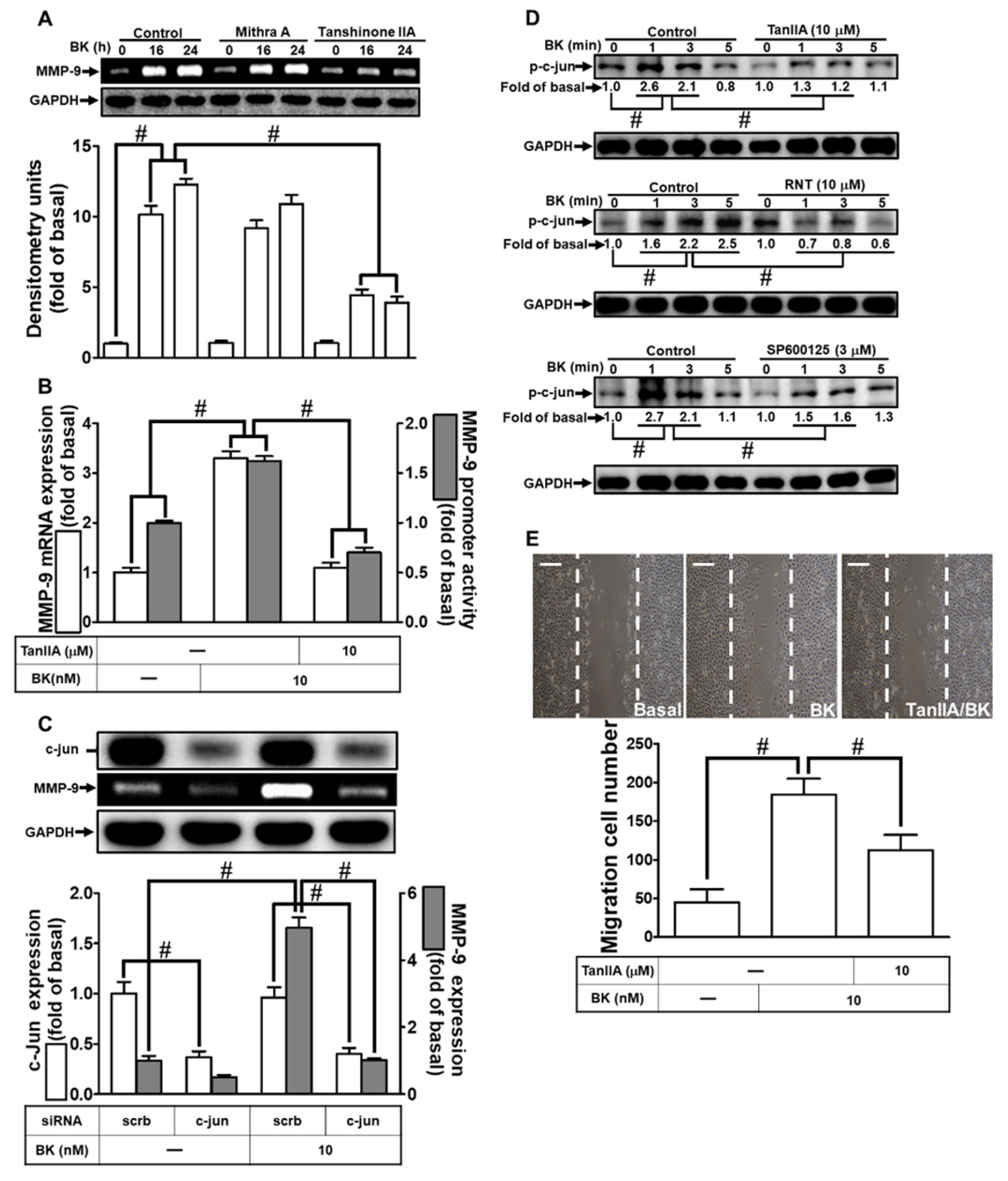
3.5. BK-Induced MMP-9 Up-Regulation in RBA-1 Cells Is Mediated through JNK1/2 Activation
3.6. RNT Suppresses BK-Stimulated MMP-9 Expression in RBA-1 Cells by Inhibiting c-Jun Activation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mugisho, O.O.; Robilliard, L.D.; Nicholson, L.F.B.; Graham, E.S.; O’Carroll, S.J. Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol. Int. 2020, 44, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Katayama, Y.; Kashiwagi, F.; Terashi, A. The role of bradykinin in mediating ischemic brain edema in rats. Stroke 1993, 24, 571–575; discussion 575–576. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K. Role of bradykinin and catecholamines in cerebral infarction and brain edema. Stroke 2009, 40, e103, author reply e104. [Google Scholar] [CrossRef]
- Chao, H.; Liu, Y.; Lin, C.; Xu, X.; Li, Z.; Bao, Z.; Fan, L.; Tao, C.; Zhao, L.; Liu, Y.; et al. Activation of bradykinin B2 receptor induced the inflammatory responses of cytosolic phospholipase A2 after the early traumatic brain injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2957–2971. [Google Scholar] [CrossRef] [PubMed]
- Francel, P.C. Bradykinin and neuronal injury. J. Neurotrauma 1992, 9 (Suppl. S1), S27–S45. [Google Scholar]
- Wang, G.; Sun, J.; Liu, G.; Fu, Y.; Zhang, X. Bradykinin promotes cell proliferation, migration, invasion, and tumor growth of gastric cancer through ERK signaling pathway. J. Cell. Biochem. 2017, 118, 4444–4453. [Google Scholar] [CrossRef]
- Liou, C.J.; Yang, C.M.; Lee, T.H.; Liu, P.S.; Hsieh, H.L. Neuroprotective effects of dehydroepiandrosterone sulfate through inhibiting expression of matrix metalloproteinase-9 from bradykinin-challenged astroglia. Mol. Neurobiol. 2019, 56, 736–747. [Google Scholar] [CrossRef]
- Lin, C.C.; Hsieh, H.L.; Shih, R.H.; Chi, P.L.; Cheng, S.E.; Chen, J.C.; Yang, C.M. NADPH oxidase 2-derived reactive oxygen species signal contributes to bradykinin-induced matrix metalloproteinase-9 expression and cell migration in brain astrocytes. Cell Commun. Signal. 2012, 10, 35. [Google Scholar] [CrossRef]
- Lee, T.H.; Liu, P.S.; Tsai, M.M.; Chen, J.L.; Wang, S.J.; Hsieh, H.L. The COX-2-derived PGE2 autocrine contributes to bradykinin-induced matrix metalloproteinase-9 expression and astrocytic migration via STAT3 signaling. Cell Commun. Signal. 2020, 18, 185. [Google Scholar] [CrossRef]
- Ramos-Fernandez, M.; Bellolio, M.F.; Stead, L.G. Matrix metalloproteinase-9 as a marker for acute ischemic stroke: A systematic review. J. Stroke Cerebrovasc. Dis. 2011, 20, 47–54. [Google Scholar] [CrossRef]
- Lorenzl, S.; Albers, D.S.; Relkin, N.; Ngyuen, T.; Hilgenberg, S.L.; Chirichigno, J.; Cudkowicz, M.E.; Beal, M.F. Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer’s disease. Neurochem. Int. 2003, 43, 191–196. [Google Scholar] [CrossRef]
- Huang, H. Matrix metalloproteinase-9 (MMP-9) as a cancer biomarker and MMP-9 biosensors: Recent advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef] [PubMed]
- Ende, C.; Gebhardt, R. Inhibition of matrix metalloproteinase-2 and -9 activities by selected flavonoids. Planta Med. 2004, 70, 1006–1008. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.J.; Karadeniz, F.; Oh, J.H.; Yu, G.H.; Jang, M.S.; Nam, K.H.; Seo, Y.; Kong, C.S. MMP-inhibitory effects of flavonoid glycosides from edible medicinal halophyte Limonium tetragonum. Evid. Based Complement. Altern. Med. 2017, 2017, 6750274. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Tsai, P.H.; Lin, C.Y.; Cheng, C.H.; Lin, T.H.; Lee, K.P.; Huang, K.Y.; Chen, S.H.; Hwang, J.J.; Kandaswami, C.C.; et al. Impact of flavonoids on matrix metalloproteinase secretion and invadopodia formation in highly invasive A431-III cancer cells. PLoS ONE 2013, 8, e71903. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Hou, W.C.; Shen, S.C.; Juan, S.H.; Ko, C.H.; Wang, L.M.; Chen, Y.C. Quercetin inhibition of tumor invasion via suppressing PKCδ/ERK/AP-1-dependent matrix metalloproteinase-9 activation in breast carcinoma cells. Carcinogenesis 2008, 29, 1807–1815. [Google Scholar] [CrossRef]
- Majtan, J.; Bohova, J.; Garcia-Villalba, R.; Tomas-Barberan, F.A.; Madakova, Z.; Majtan, T.; Majtan, V.; Klaudiny, J. Fir honeydew honey flavonoids inhibit TNF-α-induced MMP-9 expression in human keratinocytes: A new action of honey in wound healing. Arch. Dermatol. Res. 2013, 305, 619–627. [Google Scholar] [CrossRef]
- Luo, W.; Liu, Q.; Jiang, N.; Li, M.; Shi, L. Isorhamnetin inhibited migration and invasion via suppression of Akt/ERK-mediated epithelial-to-mesenchymal transition (EMT) in A549 human non-small-cell lung cancer cells. Biosci. Rep. 2019, 39, BSR20190159. [Google Scholar] [CrossRef]
- Yang, J.H.; Shin, B.Y.; Han, J.Y.; Kim, M.G.; Wi, J.E.; Kim, Y.W.; Cho, I.J.; Kim, S.C.; Shin, S.M.; Ki, S.H. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol. Appl. Pharmacol. 2014, 274, 293–301. [Google Scholar] [CrossRef]
- Lee, K.P.; Kim, J.E.; Park, W.H. Cytoprotective effect of rhamnetin on miconazole-induced H9c2 cell damage. Nutr. Res. Pract. 2015, 9, 586–591. [Google Scholar] [CrossRef]
- Pandey, A.K.; Bhattacharya, P.; Paul, S.; Patnaik, R. Rhamnetin attenuates oxidative stress and matrix metalloproteinase in animal model of ischemia/reperfusion: A possible antioxidant therapy in stroke. Am. J. Neuroprotection Neuroregeneration 2013, 5, 49–55. [Google Scholar] [CrossRef]
- Lan, L.; Wang, Y.; Pan, Z.; Wang, B.; Yue, Z.; Jiang, Z.; Li, L.; Wang, C.; Tang, H. Rhamnetin induces apoptosis in human breast cancer cells via the miR-34a/Notch-1 signaling pathway. Oncol. Lett. 2019, 17, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Kang, J.C.; Jang, Y.C.; Park, J.S.; Jang, S.Y.; Kim, D.E.; Kim, B.; Shin, H.S. Cardioprotective effects of rhamnetin in H9c2 cardiomyoblast cells under H2O2-induced apoptosis. J. Ethnopharmacol. 2014, 153, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, B.; Guo, Y.; Bai, Y.; Wang, T.; Fu, K.; Sun, G. Rhamnetin attenuates cognitive deficit and inhibits hippocampal inflammatory response and oxidative stress in rats with traumatic brain injury. Cent. Eur. J. Immunol. 2015, 40, 35–41. [Google Scholar] [CrossRef]
- Jou, T.C.; Jou, M.J.; Chen, J.Y.; Lee, S.Y. Properties of rat brain astrocytes in long-term culture. Taiwan Yi Xue Hui Za Zhi 1985, 84, 865–881. [Google Scholar] [PubMed]
- Yang, C.C.; Lin, C.C.; Jou, M.J.; Hsiao, L.D.; Yang, C.M. RTA 408 inhibits interleukin-1β-induced MMP-9 expression via suppressing protein kinase-dependent NF-κB and AP-1 activation in rat brain astrocytes. Int. J. Mol. Sci. 2019, 20, 2826. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W.; Akool, E.-S.; Rebhan, J.; Frank, S.; Beck, K.F.; Franzen, R.; Hamada, F.M.; Pfeilschifter, J. Inhibition of cytokine-induced matrix metalloproteinase 9 expression by peroxisome proliferator-activated receptor α agonists is indirect and due to a NO-mediated reduction of mRNA stability. J. Biol. Chem. 2002, 277, 33518–33528. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsiao, L.D.; Tseng, H.C.; Kuo, C.M.; Yang, C.M. Pristimerin inhibits MMP-9 expression and cell migration through attenuating NOX/ROS-dependent NF-κB activation in rat brain astrocytes challenged with LPS. J. Inflamm. Res. 2020, 13, 325–341. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsiao, L.D.; Yang, C.M. Galangin inhibits LPS-induced MMP-9 expression via suppressing protein kinase-dependent AP-1 and FoxO1 activation in rat brain astrocytes. J. Inflamm. Res. 2020, 13, 945–960. [Google Scholar] [CrossRef]
- Yong, V.W.; Forsyth, P.A.; Bell, R.; Krekoski, C.A.; Edwards, D.R. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 1998, 21, 75–80. [Google Scholar] [CrossRef]
- Yang, C.M.; Yang, S.H.; Lee, T.H.; Fang, J.Y.; Lin, C.F.; Jou, M.J.; Hsieh, H.L. Evaluation of anti-inflammatory effects of Helminthostachys zeylanica extracts via inhibiting bradykinin-induced MMP-9 expression in brain astrocytes. Mol. Neurobiol. 2016, 53, 5995–6005. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Lin, C.-C.; Hsiao, L.-D.; Kuo, J.-M.; Tseng, H.-C.; Yang, C.-M. Lipopolysaccharide-induced matrix metalloproteinase-9 expression associated with cell migration in rat brain astrocytes. Int. J. Mol. Sci. 2020, 21, 259. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Yang, C.M.; Lin, C.C. Thrombin enhanced matrix metalloproteinase-9 expression and migration of SK-N-SH cells via PAR-1, c-Src, Pyk2, EGFR, Erk1/2 and AP-1. Mol. Neurobiol. 2017, 54, 3476–3491. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yao, R.; Liu, Y.; Wang, Z.; Huang, Z.; Du, B.; Zhang, D.; Wu, L.; Xiao, L.; Zhang, Y. Isorhamnetin protects against cardiac hypertrophy through blocking PI3K–AKT pathway. Mol. Cell. Biochem. 2017, 429, 167–177. [Google Scholar] [CrossRef]
- Sherrin, T.; Blank, T.; Todorovic, C. c-Jun N-terminal kinases in memory and synaptic plasticity. Rev. Neurosci. 2011, 22, 403–410. [Google Scholar] [CrossRef]
- Raivich, G.; Behrens, A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog. Neurobiol. 2006, 78, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Galangin inhibits thrombin-induced MMP-9 expression in SK-N-SH cells via protein kinase-dependent NF-κB phosphorylation. Int. J. Mol. Sci. 2018, 19, 4084. [Google Scholar] [CrossRef]
- Yang, C.M.; Hsieh, H.L.; Yu, P.H.; Lin, C.C.; Liu, S.W. IL-1β induces MMP-9-dependent brain astrocytic migration via transactivation of PDGF receptor/NADPH oxidase 2-derived reactive oxygen species signals. Mol. Neurobiol. 2015, 52, 303–317. [Google Scholar] [CrossRef]
- Murthy, S.; Ryan, A.; He, C.; Mallampalli, R.K.; Carter, A.B. Rac1-mediated mitochondrial H2O2 generation regulates MMP-9 gene expression in macrophages via inhibition of SP-1 and AP-1. J. Biol. Chem. 2010, 285, 25062–25073. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Sato, H.; Furukawa, M.; Pagano, J.S. The expression of matrix metalloproteinase 9 is enhanced by Epstein–Barr virus latent membrane protein 1. Proc. Natl. Acad. Sci. USA 1998, 95, 3621–3626. [Google Scholar] [CrossRef]
- Ingraham, C.A.; Cox, M.E.; Ward, D.C.; Fults, D.W.; Maness, P.F. C-src and other proto-oncogenes implicated in neuronal differentiation. Mol. Chem. Neuropathol. 1989, 10, 1–14. [Google Scholar] [CrossRef]
- Girault, J.A.; Costa, A.; Derkinderen, P.; Studler, J.M.; Toutant, M. FAK and PYK2/CAKβ in the nervous system: A link between neuronal activity, plasticity and survival? Trends Neurosci. 1999, 22, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef]
- Socodato, R.; Portugal, C.C.; Canedo, T.; Rodrigues, A.; Almeida, T.O.; Henriques, J.F.; Vaz, S.H.; Magalhães, J.; Silva, C.M.; Baptista, F.I.; et al. Microglia dysfunction caused by the loss of Rhoa disrupts neuronal physiology and leads to neurodegeneration. Cell Rep. 2020, 31, 107796. [Google Scholar] [CrossRef] [PubMed]
- Salazar, S.V.; Cox, T.O.; Lee, S.; Brody, A.H.; Chyung, A.S.; Haas, L.T.; Strittmatter, S.M. Alzheimer’s disease risk factor Pyk2 mediates amyloid-β-induced synaptic dysfunction and loss. J. Neurosci. 2019, 39, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Saud, S.M.; Young, M.R.; Jones-Hall, Y.L.; Ileva, L.; Evbuomwan, M.O.; Wise, J.; Colburn, N.H.; Kim, Y.S.; Bobe, G. Chemopreventive activity of plant flavonoid isorhamnetin in colorectal cancer is mediated by oncogenic Src and β-catenin. Cancer Res. 2013, 73, 5473–5484. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Periyasamy, P.; Thangaraj, A.; Chivero, E.T.; Buch, S. PDGF/PDGFR axis in the neural systems. Mol. Aspects Med. 2018, 62, 63–74. [Google Scholar] [CrossRef]
- Fu, C.; Li, B.; Sun, Y.; Ma, G.; Yao, Y. Bradykinin inhibits oxidative stress-induced senescence of endothelial progenitor cells through the B2R/AKT/RB and B2R/EGFR/RB signal pathways. Oncotarget 2015, 6, 24675–24689. [Google Scholar] [CrossRef]
- Li, Y.; Sato, T. Dual signaling via protein kinase C and phosphatidylinositol 3′-kinase/Akt contributes to bradykinin B2 receptor-induced cardioprotection in guinea pig hearts. J. Mol. Cell Cardiol. 2001, 33, 2047–2053. [Google Scholar] [CrossRef]
- O’Shea, E.; Urrutia, A.; Green, A.R.; Colado, M.I. Current preclinical studies on neuroinflammation and changes in blood–brain barrier integrity by MDMA and methamphetamine. Neuropharmacology 2014, 87, 125–134. [Google Scholar] [CrossRef]
- Lin, C.C.; Hsieh, H.L.; Liu, S.W.; Tseng, H.C.; Hsiao, L.D.; Yang, C.M. BK induces cPLA2 expression via an autocrine loop involving COX-2-derived PGE2 in rat brain astrocytes. Mol. Neurobiol. 2015, 51, 1103–1115. [Google Scholar] [CrossRef]
- Jnawali, H.N.; Lee, E.; Jeong, K.W.; Shin, A.; Heo, Y.S.; Kim, Y. Anti-inflammatory activity of rhamnetin and a model of its binding to c-Jun NH2-terminal kinase 1 and p38 MAPK. J. Nat. Prod. 2014, 77, 258–263. [Google Scholar] [CrossRef]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wagner, E.F. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann. Rheum. Dis. 2011, 70 (Suppl. S1), i109–i112. [Google Scholar] [CrossRef]
- Chen, T.L.; Zhu, G.L.; Wang, J.A.; Zhang, G.D.; Liu, H.F.; Chen, J.R.; Wang, Y.; He, X.L. Protective effects of isorhamnetin on apoptosis and inflammation in TNF-α-induced HUVECs injury. Int. J. Clin. Exp. Pathol. 2015, 8, 2311–2320. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-M.; Lee, I.-T.; Hsiao, L.-D.; Yu, Z.-Y.; Yang, C.-C. Rhamnetin Prevents Bradykinin-Induced Expression of Matrix Metalloproteinase-9 in Rat Brain Astrocytes by Suppressing Protein Kinase-Dependent AP-1 Activation. Biomedicines 2023, 11, 3198. https://doi.org/10.3390/biomedicines11123198
Yang C-M, Lee I-T, Hsiao L-D, Yu Z-Y, Yang C-C. Rhamnetin Prevents Bradykinin-Induced Expression of Matrix Metalloproteinase-9 in Rat Brain Astrocytes by Suppressing Protein Kinase-Dependent AP-1 Activation. Biomedicines. 2023; 11(12):3198. https://doi.org/10.3390/biomedicines11123198
Chicago/Turabian StyleYang, Chuen-Mao, I-Ta Lee, Li-Der Hsiao, Zih-Yao Yu, and Chien-Chung Yang. 2023. "Rhamnetin Prevents Bradykinin-Induced Expression of Matrix Metalloproteinase-9 in Rat Brain Astrocytes by Suppressing Protein Kinase-Dependent AP-1 Activation" Biomedicines 11, no. 12: 3198. https://doi.org/10.3390/biomedicines11123198