

Networking to Optimize Dmd exon 53 Skipping in the Brain of mdx52 Mouse Model

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
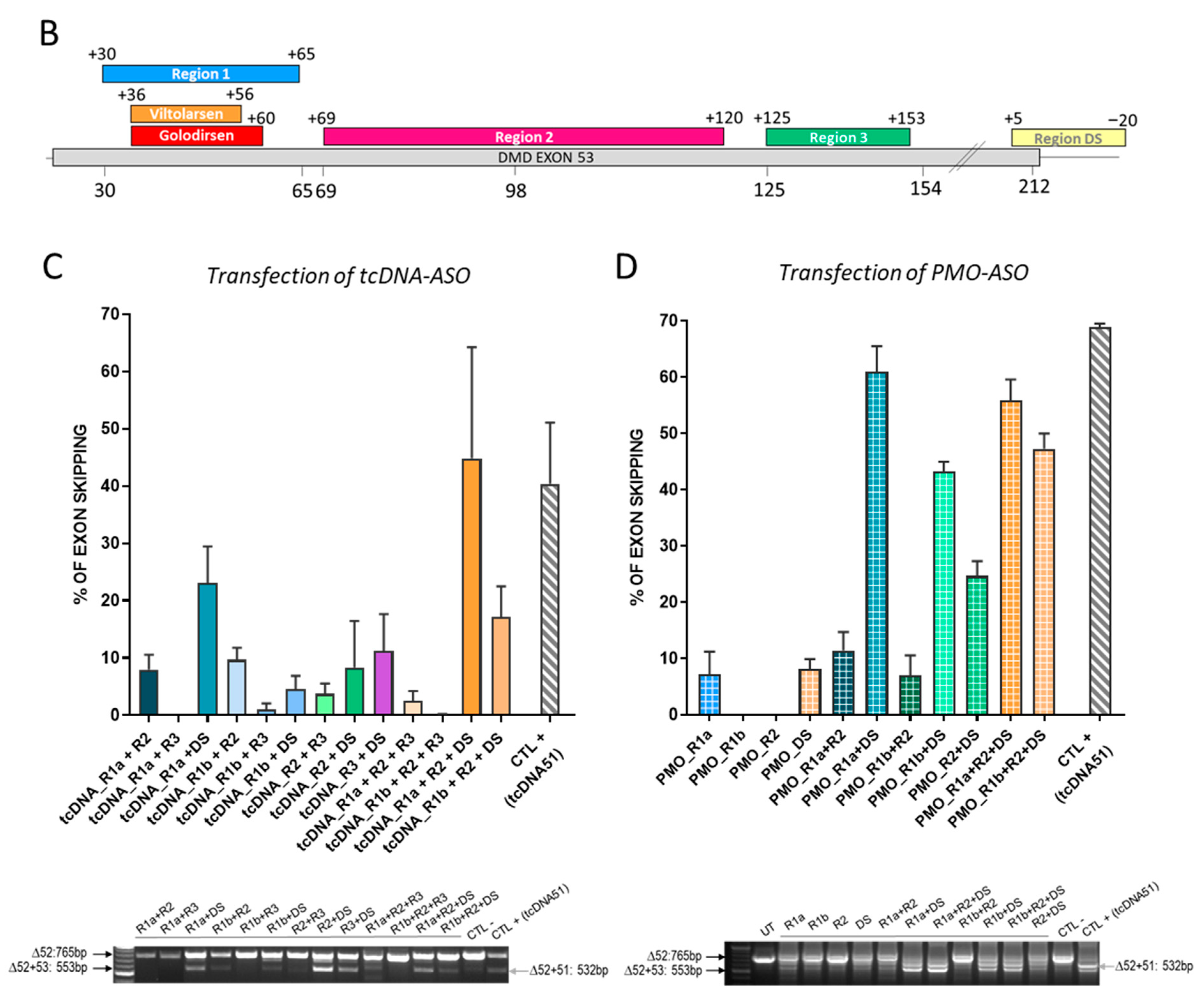
3.1. In Vitro Screening of ASO Sequences Targeting Mouse Dmd Exon 53
3.2. Intramuscular Injection of Single or Combined ASO Targeting Exon 53 in mdx52 Mice
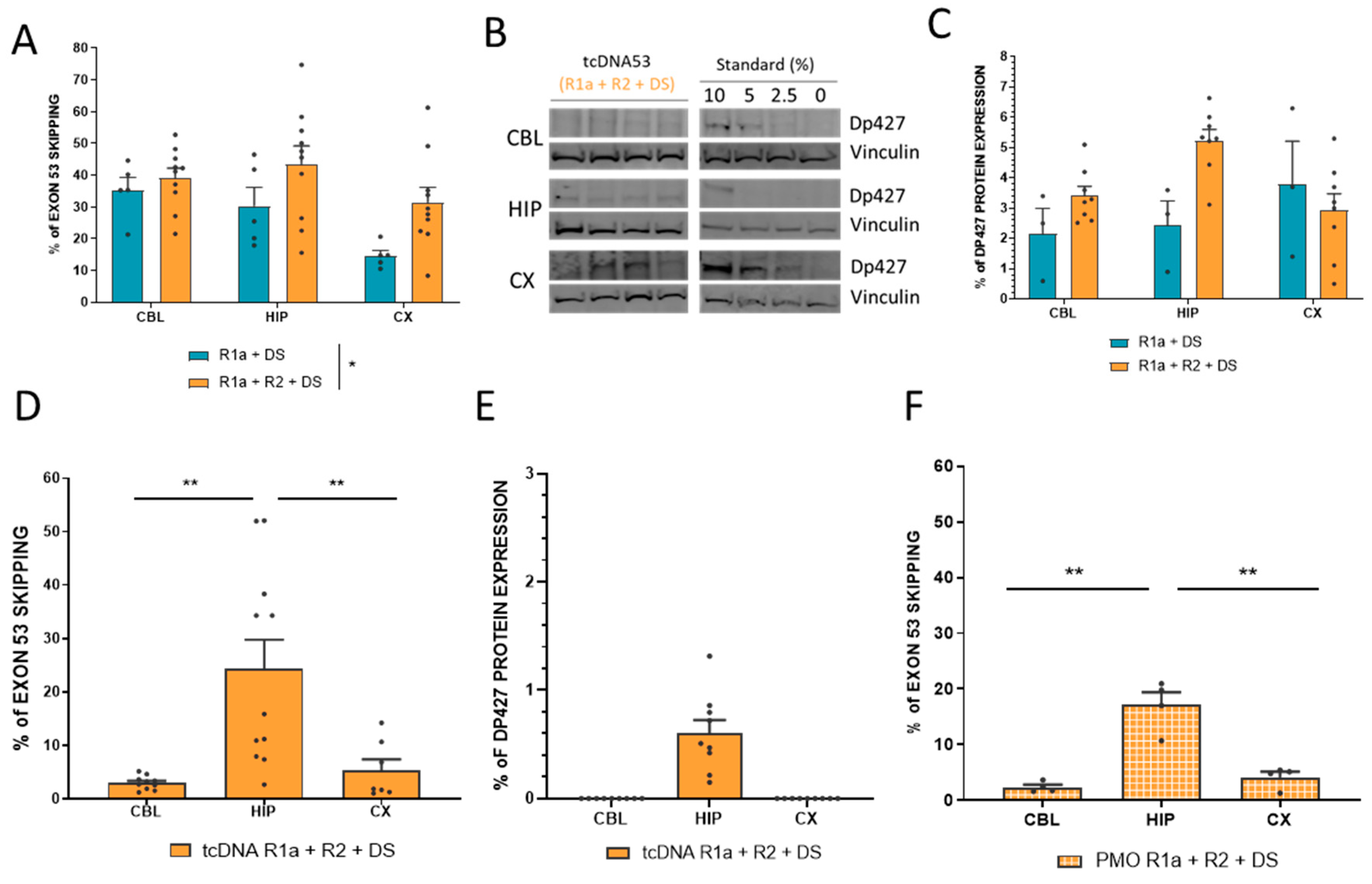
3.3. Intracerebroventricular Injection of Combined ASOs Targeting Exon 53 in mdx52 Mice
3.4. Combination of four ASOs Targeting Exon 53 Skipping Achieves Higher Level of Exon Skipping but Still Only Very Low Levels of Protein Restoration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colombo, P.; Nobile, M.; Tesei, A.; Civati, F.; Gandossini, S.; Mani, E.; Molteni, M.; Bresolin, N.; D’Angelo, G. Assessing Mental Health in Boys with Duchenne Muscular Dystrophy: Emotional, Behavioural and Neurodevelopmental Profile in an Italian Clinical Sample. Eur. J. Paediatr. Neurol. 2017, 21, 639–647. [Google Scholar] [CrossRef]
- Hinton, V.J.; Cyrulnik, S.E.; Fee, R.J.; Batchelder, A.; Kiefel, J.M.; Goldstein, E.M.; Kaufmann, P.; De Vivo, D.C. Association of Autistic Spectrum Disorders with Dystrophinopathies. Pediatr. Neurol. 2009, 41, 339–346. [Google Scholar] [CrossRef]
- Maresh, K.; Papageorgiou, A.; Ridout, D.; Harrison, N.A.; Mandy, W.; Skuse, D.; Muntoni, F. Startle Responses in Duchenne Muscular Dystrophy: A Novel Biomarker of Brain Dystrophin Deficiency. Brain 2023, 146, 252–265. [Google Scholar] [CrossRef]
- Ricotti, V.; Mandy, W.P.L.; Scoto, M.; Pane, M.; Deconinck, N.; Messina, S.; Mercuri, E.; Skuse, D.H.; Muntoni, F. Neurodevelopmental, Emotional, and Behavioural Problems in Duchenne Muscular Dystrophy in Relation to Underlying Dystrophin Gene Mutations. Dev. Med. Child Neurol. 2016, 58, 77–84. [Google Scholar] [CrossRef]
- Mullard, A. FDA Approves First Gene Therapy for Duchenne Muscular Dystrophy, despite Internal Objections. Nat. Rev. Drug Discov. 2023, 22, 610. [Google Scholar] [CrossRef] [PubMed]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon Skipping Leading to an Artificial DMD Protein Lacking Amino Acids from Exons 45 through 55 Could Rescue up to 63% of Patients with Duchenne Muscular Dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Nakamura, K.; Nakao, K.; Kameya, S.; Kobayashi, O.; Nonaka, I.; Kobayashi, T.; Katsuki, M. Targeted Disruption of Exon 52 in the Mouse Dystrophin Gene Induced Muscle Degeneration Similar to That Observed in Duchenne Muscular Dystrophy. Biochem. Biophys. Res. Commun. 1997, 238, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Felisari, G.; Martinelli Boneschi, F.; Bardoni, A.; Sironi, M.; Comi, G.P.; Robotti, M.; Turconi, A.C.; Lai, M.; Corrao, G.; Bresolin, N. Loss of Dp140 Dystrophin Isoform and Intellectual Impairment in Duchenne Dystrophy. Neurology 2000, 55, 559–564. [Google Scholar] [CrossRef]
- Hendriksen, J.G.M.; Thangarajh, M.; Kan, H.E.; Muntoni, F. ENMC 249th workshop study group 249th ENMC International Workshop: The Role of Brain Dystrophin in Muscular Dystrophy: Implications for Clinical Care and Translational Research, Hoofddorp, The Netherlands, November 29th-December 1st 2019. Neuromuscul. Disord. 2020, 30, 782–794. [Google Scholar] [CrossRef]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Jiménez-López, E.; Saz-Lara, A.; Martínez-García, I.; Martínez-Vizcaíno, V. Global Prevalence of Intellectual Developmental Disorder in Dystrophinopathies: A Systematic Review and Meta-Analysis. Dev. Med. Child. Neurol. 2023, 65, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Betts, G.A.; Maroulis, S.; Gilissen, C.; Pedersen, R.L.; Mowat, D.R.; Johnston, H.M.; Buckley, M.F. Dystrophin Gene Mutation Location and the Risk of Cognitive Impairment in Duchenne Muscular Dystrophy. PLoS ONE 2010, 5, e8803. [Google Scholar] [CrossRef] [PubMed]
- Saoudi, A.; Barberat, S.; le Coz, O.; Vacca, O.; Doisy Caquant, M.; Tensorer, T.; Sliwinski, E.; Garcia, L.; Muntoni, F.; Vaillend, C.; et al. Partial Restoration of Brain Dystrophin by Tricyclo-DNA Antisense Oligonucleotides Alleviates Emotional Deficits in Mdx52 Mice. Mol. Ther. Nucleic Acids 2023, 32, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Saoudi, A.; Fergus, C.; Gileadi, T.; Montanaro, F.; Morgan, J.E.; Kelly, V.P.; Tensorer, T.; Garcia, L.; Vaillend, C.; Muntoni, F.; et al. Investigating the Impact of Delivery Routes for Exon Skipping Therapies in the CNS of DMD Mouse Models. Cells 2023, 12, 908. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Yokota, T.; Nagata, T.; Nakamura, A.; Tanihata, J.; Saito, T.; Duguez, S.M.R.; Nagaraju, K.; Hoffman, E.P.; Partridge, T.; et al. Bodywide Skipping of Exons 45-55 in Dystrophic Mdx52 Mice by Systemic Antisense Delivery. Proc. Natl. Acad. Sci. USA 2012, 109, 13763–13768. [Google Scholar] [CrossRef]
- Blau, H.M.; Chiu, C.P.; Webster, C. Cytoplasmic Activation of Human Nuclear Genes in Stable Heterocaryons. Cell 1983, 32, 1171–1180. [Google Scholar] [CrossRef]
- Relizani, K.; Echevarría, L.; Zarrouki, F.; Gastaldi, C.; Dambrune, C.; Aupy, P.; Haeberli, A.; Komisarski, M.; Tensorer, T.; Larcher, T.; et al. Palmitic Acid Conjugation Enhances Potency of Tricyclo-DNA Splice Switching Oligonucleotides. Nucleic Acids Res. 2022, 50, 17–34. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: San Diego, CA, USA, 2001; ISBN 978-0-12-547636-2. [Google Scholar]
- Aupy, P.; Zarrouki, F.; Sandro, Q.; Gastaldi, C.; Buclez, P.-O.; Mamchaoui, K.; Garcia, L.; Vaillend, C.; Goyenvalle, A. Long-Term Efficacy of AAV9-U7snRNA-Mediated Exon 51 Skipping in Mdx52 Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 1037–1047. [Google Scholar] [CrossRef]
- Mitrpant, C.; Adams, A.M.; Meloni, P.L.; Muntoni, F.; Fletcher, S.; Wilton, S.D. Rational Design of Antisense Oligomers to Induce Dystrophin Exon Skipping. Mol. Ther. 2009, 17, 1418–1426. [Google Scholar] [CrossRef]
- Saoudi, A.; Zarrouki, F.; Sebrié, C.; Izabelle, C.; Goyenvalle, A.; Vaillend, C. Emotional Behavior and Brain Anatomy of the Mdx52 Mouse Model of Duchenne Muscular Dystrophy. Dis. Model. Mech. 2021, 14, dmm049028. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Kaman, W.E.; Weij, R.; den Dunnen, J.T.; van Ommen, G.J.; van Deutekom, J.C. Exploring the Frontiers of Therapeutic Exon Skipping for Duchenne Muscular Dystrophy by Double Targeting within One or Multiple Exons. Mol. Ther. 2006, 14, 401–407. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sivetz, N.; Layne, J.; Voss, D.M.; Yang, L.; Zhang, Q.; Krainer, A.R. Exon-Skipping Antisense Oligonucleotides for Cystic Fibrosis Therapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2114858118. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Aoki, Y.; Miskew, B.; Panesar, D.; Touznik, A.; Nagata, T.; Tanihata, J.; Nakamura, A.; Nagaraju, K.; Yokota, T. Long-Term Efficacy of Systemic Multiexon Skipping Targeting Dystrophin Exons 45-55 with a Cocktail of Vivo-Morpholinos in Mdx52 Mice. Mol. Ther. Nucleic Acids 2015, 4, e225. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45-55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol. Ther. 2019, 27, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; van den Bergen, J.C.; Verhaart, I.E.C.; Wokke, B.; Janson, A.A.M.; van den Eijnde, R.; den Dunnen, J.T.; Laros, J.F.J.; Verschuuren, J.J.G.M.; ’t Hoen, P.A.C.; et al. DMD Transcript Imbalance Determines Dystrophin Levels. FASEB J. 2013, 27, 4909–4916. [Google Scholar] [CrossRef]
- García-Rodríguez, R.; Hiller, M.; Jiménez-Gracia, L.; van der Pal, Z.; Balog, J.; Adamzek, K.; Aartsma-Rus, A.; Spitali, P. Premature Termination Codons in the DMD Gene Cause Reduced Local mRNA Synthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 16456–16464. [Google Scholar] [CrossRef]
- Clemens, P.R.; Rao, V.K.; Connolly, A.M.; Harper, A.D.; Mah, J.K.; Smith, E.C.; McDonald, C.M.; Zaidman, C.M.; Morgenroth, L.P.; Osaki, H.; et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 982–991. [Google Scholar] [CrossRef]
- Kudoh, H.; Ikeda, H.; Kakitani, M.; Ueda, A.; Hayasaka, M.; Tomizuka, K.; Hanaoka, K. A New Model Mouse for Duchenne Muscular Dystrophy Produced by 2.4 Mb Deletion of Dystrophin Gene Using Cre-loxP Recombination System. Biochem. Biophys. Res. Commun. 2005, 328, 507–516. [Google Scholar] [CrossRef]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.J.G.M.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and Localization of Human Dystrophin Isoform Expression Provide Insights into the Cognitive Phenotype of Duchenne Muscular Dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kuniishi, H.; Sakai, K.; Fukushima, Y.; Du, X.; Yamashiro, K.; Hori, K.; Imamura, M.; Hoshino, M.; Yamada, M.; et al. Brain Dp140 Alters Glutamatergic Transmission and Social Behaviour in the Mdx52 Mouse Model of Duchenne Muscular Dystrophy. Prog. Neurobiol. 2022, 216, 102288. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHEMISTRY | REGION | NAME | TARGET POSITION | ABBREVIATED NAME | TARGET SEQUENCE |
|---|---|---|---|---|---|
| tcDNA | PRE-SCREENING | tcDNA +27+41 | +27+41 | GAGGTTCAAGAACAG | |
| tcDNA +33+47 | +33+47 | CAAGAACAGCTGCAG | |||
| tcDNA +36+50 | +36+50 | GAACAGCTGCAGAAC | |||
| tcDNA +38+52 | +38+52 | ACAGCTGCAGAACAG | |||
| tcDNA +40+54 | +40+54 | AGCTGCAGAACAGGA | |||
| tcDNA +41+55 | +41+55 | GCTGCAGAACAGGAG | |||
| tcDNA +42+56 | +42+56 | CTGCAGAACAGGAGA | |||
| tcDNA +43+57 | +43+57 | TGCAGAACAGGAGAC | |||
| tcDNA +44+58 | +44+58 | GCAGAACAGGAGACA | |||
| tcDNA +46+60 | +46+60 | AGAACAGGAGACAAC | |||
| tcDNA +59+73 | +59+73 | ACAGTTGAATGAAAT | |||
| tcDNA +73+87 | +73+87 | TGTTAAAGGATTCAA | |||
| R1 | tcDNA +33+52 | +33+52 | CAAGAACAGCTGCAGAACAG | ||
| tcDNA +36+55 | +36+55 | tcDNA_R1a | GAACAGCTGCAGAACAGGAG | ||
| tcDNA +39+58 | +39+58 | CAGCTGCAGAACAGGAGACA | |||
| tcDNA +42+61 | +42+61 | CTGCAGAACAGGAGACAACA | |||
| tcDNA +45+64 | +45+64 | tcDNA_R1b | CAGAACAGGAGACAACAGTT | ||
| tcDNA +56+75 | +56+75 | ACAACAGTTGAATGAAATGT | |||
| R2 | tcDNA +69+88 | +69+88 | GAAATGTTAAAGGATTCAAC | ||
| tcDNA +72+91 | +72+91 | tcDNA_R2 | ATGTTAAAGGATTCAACACA | ||
| tcDNA +75+94 | +75+94 | TTAAAGGATTCAACACAATG | |||
| tcDNA +78+97 | +78+97 | AAGGATTCAACACAATGGCT | |||
| tcDNA +100+120 | +100+120 | AAGCTAAGGAAGAAGCCGAA | |||
| R3 | tcNDA +125+144 | +125+144 | TCATAGGACAGGTCAGAGGC | ||
| tcDNA +128+147 | +128+147 | TAGGACAGGTCAGAGGCAAG | |||
| tcDNA +131+150 | +131+150 | tcDNA_R3 | GACAGGTCAGAGGCAAGCTT | ||
| tcDNA +134+153 | +134+153 | AGGTCAGAGGCAAGCTTGAC | |||
| DS | tcDNA +5−15 | +5−15 | tcDNA_DS | CCAAGgttagtgtcaagcat | |
| tcDNA +3−18 | +3−18 | AAGgttagtgtcaagcatat | |||
| tcDNA +1-20 | +1-20 | Ggttagtgtcaagcatatct | |||
| PMO | R1 | PMO +36+60 | +36+60 | PMO_R1a | GAACAGCTGCAGAACAGGAGACAAC |
| PMO +43+67 | +43+67 | PMO_R1b | GCAGAACAGGAGACAACAGTTGAAT | ||
| R2 | PMO +69+98 | +69+98 | PMO_R2 | GAAATGTTAAAGGATTCAACACAAT | |
| R3 | No PMO for this region | ||||
| DS | PMO +9−16 | +9−16 | PMO_DS | GAAACCAAGgttagtgtcaagcata | |
| MOE | MOE +9+26 | +9+26 | TTCAGATTCAGTGGGATGTG | ||
| R1 | MOE +28+45 | +28+45 | MOE_R1 | GGTTCAAGAACAGCTGCA | |
| R2 | MOE +79+98 | +79+98 | MOE_R2 | TCAACACAATGGCTGGAAGCT | |
| MOE +83+102 | +83+102 | CAACACAATGGCTGGAAGCT | |||
| MOE +84+103 | +84+103 | TCAACACAATGGCTGGAAGC | |||
| R3 | MOE +135+154 | +135+154 | MOE_R3 | TCAGAGGCAAGCTTGACTCA | |
| DS | MOE +5−15 | +5−15 | MOE_DS | CCAAGgttagtgtcaagcat | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doisy, M.; Vacca, O.; Fergus, C.; Gileadi, T.; Verhaeg, M.; Saoudi, A.; Tensorer, T.; Garcia, L.; Kelly, V.P.; Montanaro, F.; et al. Networking to Optimize Dmd exon 53 Skipping in the Brain of mdx52 Mouse Model. Biomedicines 2023, 11, 3243. https://doi.org/10.3390/biomedicines11123243
Doisy M, Vacca O, Fergus C, Gileadi T, Verhaeg M, Saoudi A, Tensorer T, Garcia L, Kelly VP, Montanaro F, et al. Networking to Optimize Dmd exon 53 Skipping in the Brain of mdx52 Mouse Model. Biomedicines. 2023; 11(12):3243. https://doi.org/10.3390/biomedicines11123243
Chicago/Turabian StyleDoisy, Mathilde, Ophélie Vacca, Claire Fergus, Talia Gileadi, Minou Verhaeg, Amel Saoudi, Thomas Tensorer, Luis Garcia, Vincent P. Kelly, Federica Montanaro, and et al. 2023. "Networking to Optimize Dmd exon 53 Skipping in the Brain of mdx52 Mouse Model" Biomedicines 11, no. 12: 3243. https://doi.org/10.3390/biomedicines11123243