
SNPs-Panel Polymorphism Variations in GHRL and GHSR Genes Are Not Associated with Prostate Cancer
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. DNA Extraction
2.3. Genotyping
2.4. Statistical Analysis
3. Results
3.1. Association of GHRL and GHSR SNPs with PCa Risk
3.2. Analysis of GHRL and GHSR Haplotypes with PCa Risk
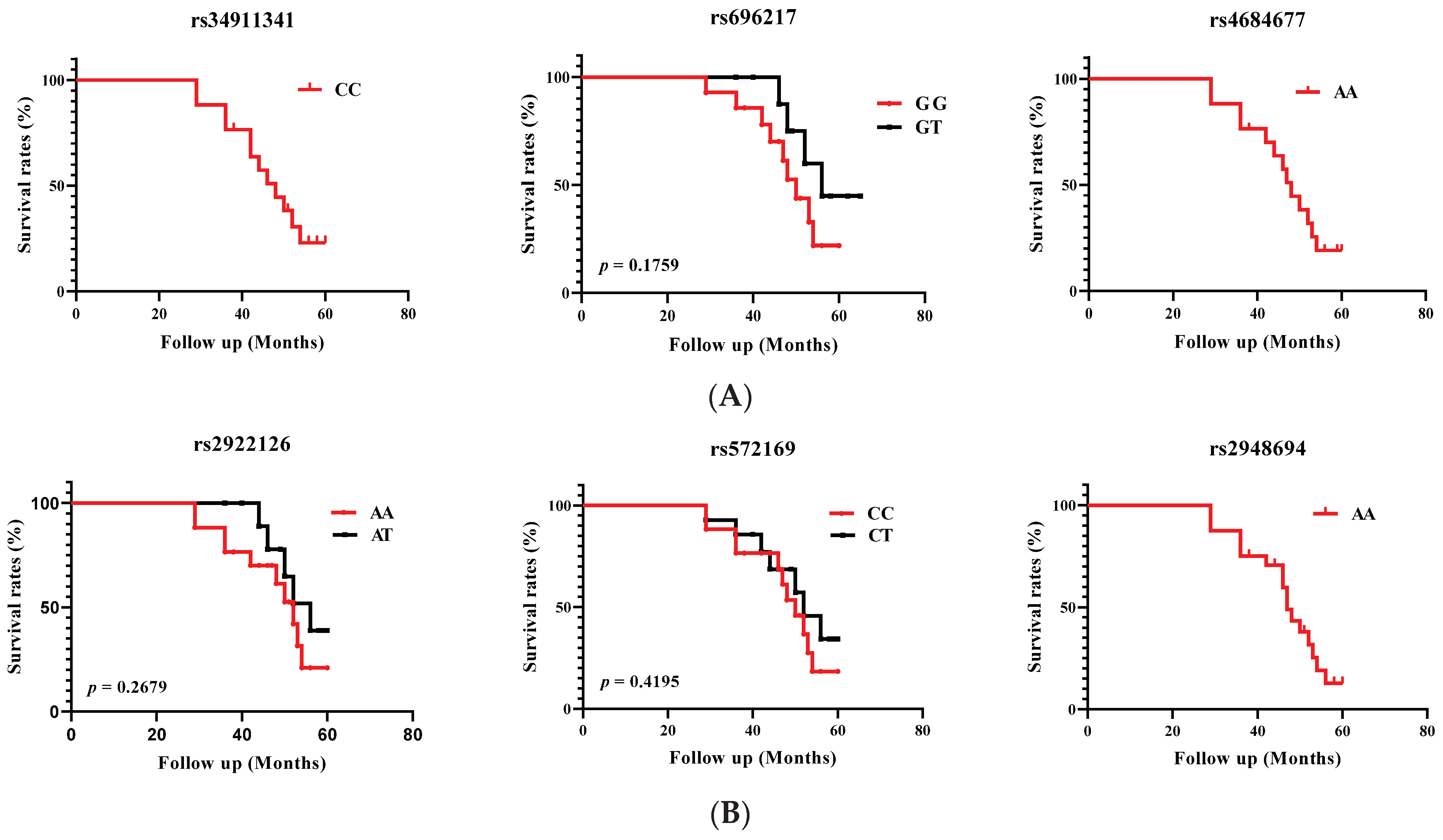
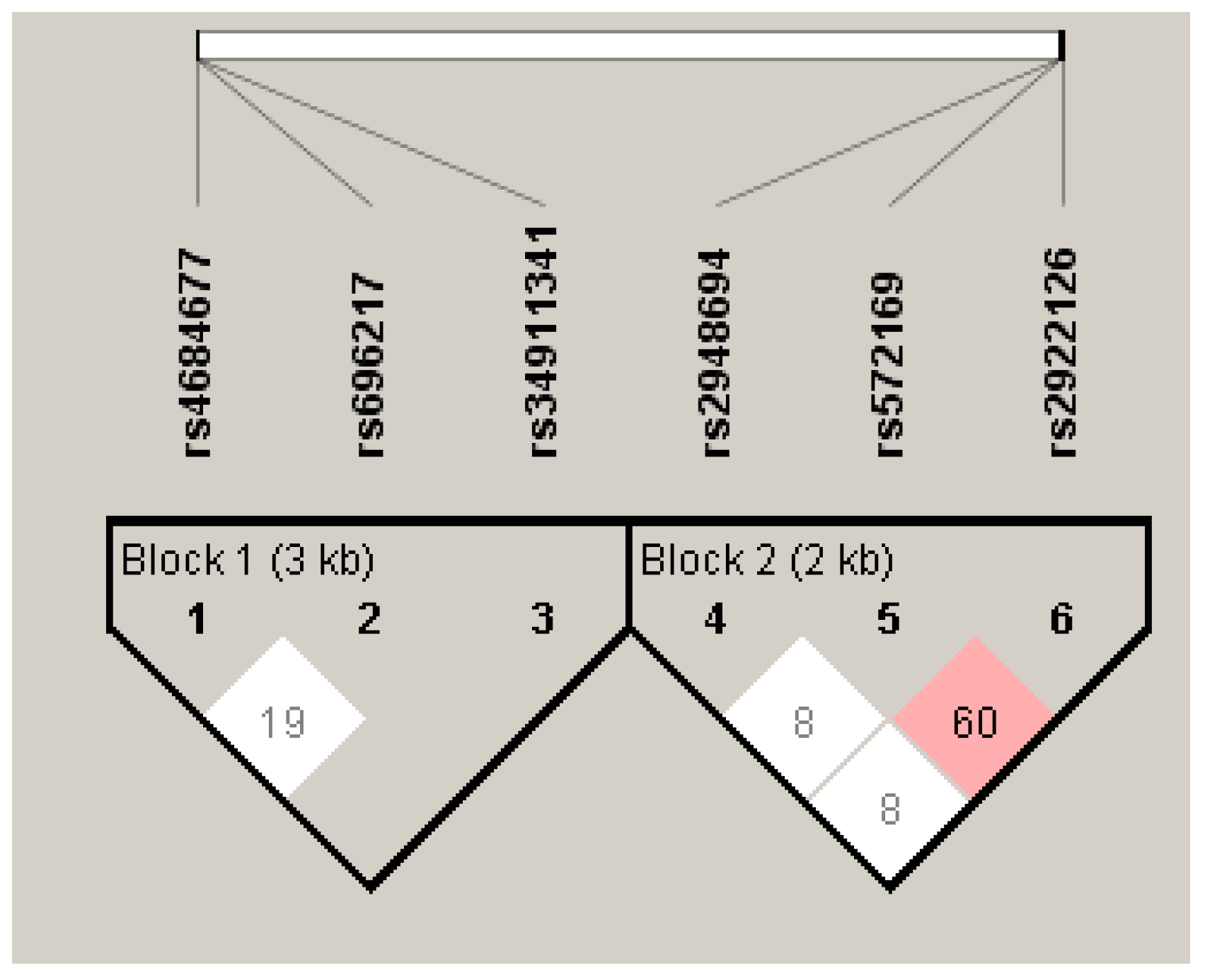
3.3. Linkage Disequilibrium Analysis of the GHRL and GHSR SNPs
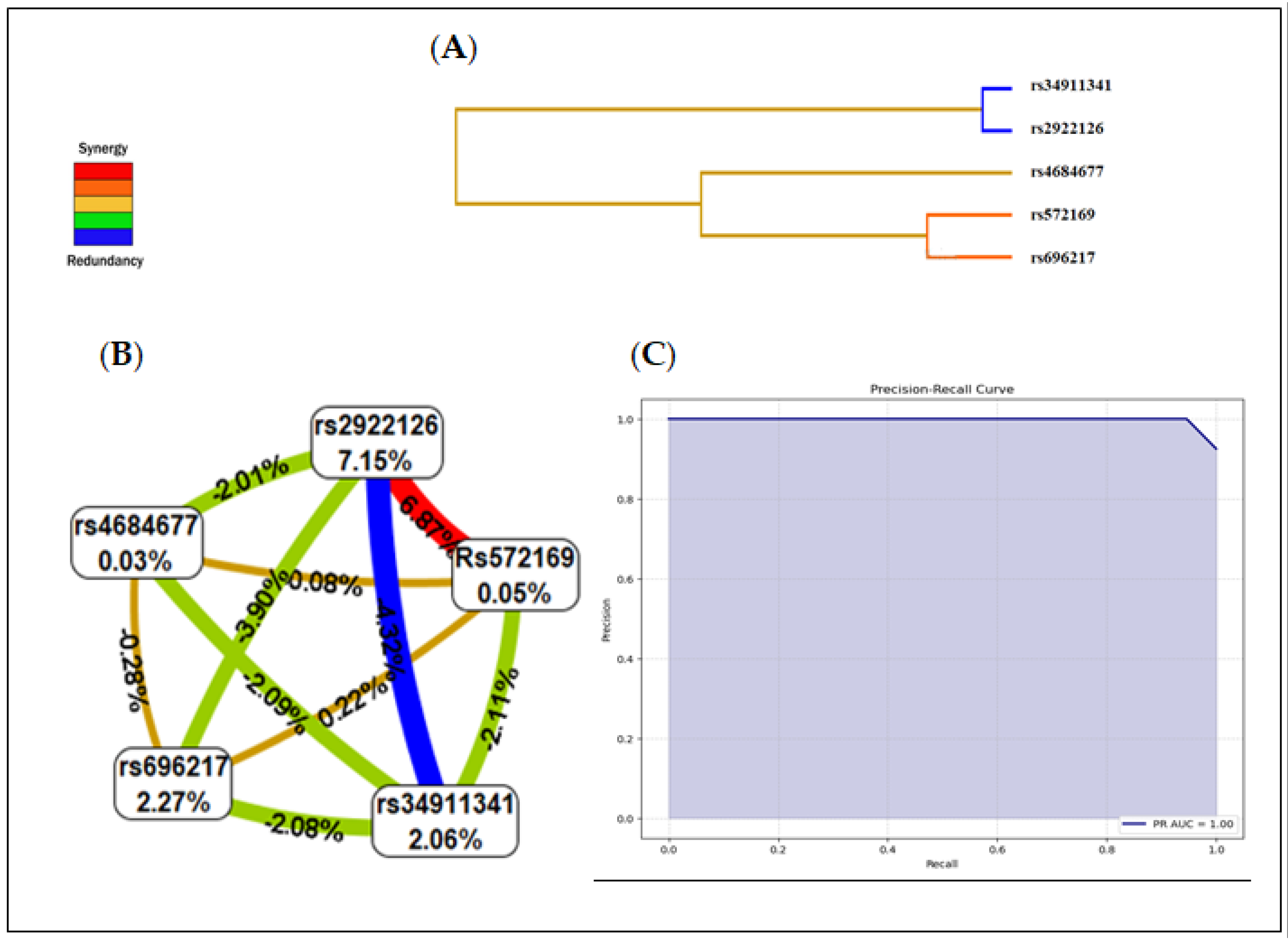
3.4. Multifactor Dimensionality Reduction (MDR)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GLOBOCAN 2020. New Global Cancer Data. Available online: www.uicc.org/news/globocan-2020-new-global-cancer-data (accessed on 23 October 2023).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Feigelson, H.S.; Goddard, K.A.; Hollombe, C.; Tingle, S.R.; Gillanders, E.M.; Mechanic, L.E.; Nelson, S.A. Approaches to integrating germline and tumor genomic data in cancer research. Carcinogenesis 2014, 35, 2157–2163. [Google Scholar] [CrossRef]
- Hicks, C.; Miele, L.; Koganti, T.; Vijayakumar, S. Comprehensive assessment and network analysis of the emerging genetic susceptibility landscape of prostate cancer. Cancer Inform. 2013, 12, 175–191. [Google Scholar] [CrossRef]
- Bourefis, A.; Berredjem, H.; Djeffal, O.; Le, T.K.; Giusiano, S.; Rocchi, P. HSP27/Menin Expression as New Prognostic Serum Biomarkers of Prostate Cancer Aggressiveness Independent of PSA. Cancers 2022, 14, 4773. [Google Scholar] [CrossRef]
- Bukhari, S.N.A. An insight into the multifunctional role of ghrelin and structure activity relationship studies of ghrelin receptor ligands with clinical trials. Eur. J. Med. Chem. 2022, 235, 114308. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Asakawa, A.; Inui, A.; Kaga, T.; Yuzuriha, H.; Nagata, T.; Ueno, N.; Makino, S.; Fujimiya, M.; Niijima, A.; Fujino, M.A.; et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology 2001, 120, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Gahete, M.D.; Rincon-Fernandez, D.; Villa-Osaba, A.; Hormaechea-Agulla, D.; Ibanez-Costa, A.; Martinez-Fuentes, A.J.; Gracia-Navarro, F.; Castaño, J.P.; Luque, R.M. Ghrelin gene products, receptors, and GOAT enzyme: Biological and pathophysiological insight. J. Endocrinol. 2014, 220, R1eR24. [Google Scholar] [CrossRef]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, T.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Lanfranco, F.; Bonelli, L.; Baldi, M.; Me, E.; Broglio, F.; Ghigo, E. Acylated ghrelin inhibits spontaneous luteinizing hormone pulsatility and responsiveness to naloxone but not that to gonadotropin-releasing hormone in young men: Evidence for a central inhibitory action of ghrelin on the gonadal axis. J. Clin. Endocrinol. Metab. 2008, 93, 3633–3639. [Google Scholar] [CrossRef]
- Kotta, A.S.; Kelling, A.S.; Corleto, K.A.; Sun, Y.; Giles, E.D. Ghrelin and Cancer: Examining the Roles of the Ghrelin Axis in Tumor Growth and Progression. Biomolecules 2022, 12, 483. [Google Scholar] [CrossRef]
- Lin, T.C.; Liu, Y.P.; Chan, Y.C.; Su, C.Y.; Lin, Y.F.; Hsu, S.L.; Yang, C.S.; Hsiao, M. Ghrelin promotes renal cell carcinoma metastasis via Snail activation and is associated with poor prognosis. J. Pathol. 2015, 237, 50–61. [Google Scholar] [CrossRef]
- Kraus, D.; Reckenbeil, J.; Wenghoefer, M.; Stark, H.; Frentzen, M.; Allam, J.P.; Novak, N.; Frede, S.; Götz, W.; Probstmeier, R. Ghrelin promotes oral tumor cell proliferation by modifying GLUT1 expression. Cell. Mol. Life Sci. 2016, 73, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, D.; Pradhan, G.; Zhou, Y.; Xue, B.; Sun, Y. Diverse and Complementary Effects of Ghrelin and Obestatin. Biomolecules 2022, 12, 517. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, M.S.; Waseem, T.; Ito, H.; Robinson, M.K.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Ghrelin promotes pancreatic adenocarcinoma cellular proliferation and invasiveness. Biochem. Biophys. Res. Commun. 2003, 309, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Yeh, A.H.; Jeffery, P.L.; Duncan, R.P.; Herington, A.C.; Chopin, L.K. Ghrelin and a novel preproghrelin isoform are highly expressed in prostate cancer and ghrelin activates mitogen-activated protein kinase in prostate cancer. Clin. Cancer Res. 2005, 11, 8295–8303. [Google Scholar] [CrossRef]
- Spiridon, I.A.; Apostol Ciobanu, D.G.; Giușcă, S.E.; Căruntu, D.I. Ghrelin and its role in gastrointestinal tract tumors (Review). Mol. Med. Rev. 2021, 24, 663. [Google Scholar] [CrossRef] [PubMed]
- Cassoni, P.; Ghé, C.; Marrocco, T.; Tarabra, E.; Allia, E.; Catapano, F.; Deghenghi, R.; Ghigo, E.; Papotti, M.; Muccioli, G. Expression of ghrelin and biological activity of specific receptors for ghrelin and des-acyl ghrelin in human prostate neoplasms and related cell lines. Eur. J. Endocrinol. 2004, 150, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Borell, H.; Gardin, A.; Ufer, M.; Huth, F.; Camenisch, G. In vitro studies and in silico predictions of fluconazole and CYP2C9 genetic polymorphism impact on siponimod metabolism and pharmacokinetics. Eur. J. Clin. Pharmacol. 2018, 74, 455–464. [Google Scholar] [CrossRef]
- Garcia, E.A.; King, P.; Sidhu, K.; Ohgusu, H.; Walley, A.; Lecoeur, C.; Gueorguiev, M.; Khalaf, S.; Davies, D.; Grossman, A.B. The role of ghrelin and ghrelin-receptor gene variants and promoter activity in type 2 diabetes. Eur. J. Endocrinol. 2009, 161, 307–315. [Google Scholar] [CrossRef]
- Liao, N.; Xie, Z.K.; Huang, J.; Xie, Z.F. Association between the ghrelin Leu72Met polymorphism and type 2 diabetes risk: A meta-analysis. Gene 2013, 517, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Gjesing, A.P.; Larsen, L.H.; Torekov, S.S.; Hainerova, I.A.; Kapur, R.; Johansen, A.; Albrechtsen, A.; Boj, S.; Holst, B.; Harper, A. Family and population-based studies of variation within the ghrelin receptor locus in relation to measures of obesity. PLoS ONE 2010, 5, e10084. [Google Scholar] [CrossRef]
- Ukkola, O.; Paakko, T.; Kesaniemi, Y.A. Ghrelin and its promoter variant associated with cardiac hypertrophy. J. Hum. Hypertens. 2011, 26, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Adam, V.; Palomera, E.; Blesa, S.; Díaz, G.; Buquet, X.; Serra-Prat, M.; Martín-Escudero, J.C.; Palanca, A.; Chaves, J.F. Ghrelin Gene Variants Influence on Metabolic Syndrome Components in Aged Spanish Population. PLoS ONE 2015, 10, e0136931. [Google Scholar] [CrossRef]
- Berthold, H.K.; Giannakidou, E.; Krone, W.; Tregouet, D.A.; Gouni-Berthold, I. Influence of ghrelin gene polymorphisms on hypertension and atherosclerotic disease. Hypertens. Res. 2010, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, M.; Kassim, S.K.; Khairy, E.; Maher, M.; Mansour, K.A.; Albreedy, A.M. Ghrelin gene polymorphism as a genetic biomarker for prediction of therapy induced clearance in Egyptian chronic HCV patients. Gene 2018, 649, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Dardennes, R.M.; Zizzari, P.; Tolle, V.; Foulon, C.; Kipma, A.; Romo, L.; Iancu-Gontard, D.; Boni, C.; Sinet, P.M.; Bluet, M.T.; et al. Family trios analysis of common polymorphisms in the obestatin/ghrelin, BDNF and AGRP genes in patients with Anorexia nervosa: Association with subtype, body-mass index, severity and age of onset. Psychoneuroendocrinology 2007, 32, 106–113. [Google Scholar] [CrossRef]
- Blum, D.; Wolf-Linder, S.; Oberholzer, R.; Brändle, M.; Hundsberger, T.; Strasser, F. Natural ghrelin in advanced cancer patients with cachexia, a case series. J. Cachexia Sarcopenia Muscle 2021, 12, 506–516. [Google Scholar] [CrossRef]
- Landgren, S.; Jerlhag, E.; Zetterberg, H.; Gonzalez-Quintela, A.; Campos, J.; Olofsson, U.; Nilsson, S.; Blennow, K.; Engel, J.A. Association of pro-ghrelin and GHS-R1A gene polymorphisms and haplotypes with heavy alcohol use and body mass. Alcohol Clin. Exp. Res. 2008, 32, 2054–2061. [Google Scholar] [CrossRef]
- Langre, S.; Berglund, K.; Jerlhag, E.; Fahlke, C.; Balldin, J.; Berggren, U.; Zetterberg, H.; Blennow, K.; Jörgen, A. Reward-Related Genes and Personality Traits in Alcohol-Dependent Individuals: A Pilot Case Control Study. Neuropsychobiology 2011, 64, 38–46. [Google Scholar] [CrossRef]
- Steinle, N.I.; Pollin, T.I.; O’Connell, J.R.; Mitchell, B.D.; Shuldiner, A.R. Variants in the ghrelin gene are associated with metabolic syndrome in the Old Order Amish. J. Clin. Endocrinol. Metab. 2005, 90, 6672–6677. [Google Scholar] [CrossRef]
- Hansson, C.; Annerbrink, K.; Nilsson, S.; Bah, J.; Olsson, M.; Allgulander, C.; Andersch, S.; Sjodin, I.; Eriksson, E.; Dickson, S.L. A possible association between panic disorder and a polymorphism in the preproghrelingene. Psychiatry Res. 2013, 206, 22–25. [Google Scholar] [CrossRef]
- Pabalan, N.A.; Seim, I.; Jarjanazi, H.; Chopin, L.K. Associations between ghrelin and ghrelin receptor polymorphisms and cancer in Caucasian populations: A meta-analysis. BMC Genet. 2014, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Moniuszko, A.; Wawrusiewicz-Kurylonek, N.; Bossowska, A.; Goscik, J.; Luczynski, W.; Glowinska-Olszewska, B.; Kretowski, A.; Bossowski, A. The association between rs4684677 T/A polymorphism in preproghrelin gene and predisposition to autoimmune thyroid diseases in children. Autoimmunity 2015, 48, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Doecke, J.D.; Zhao, Z.Z.; Stark, M.S.; Green, A.C.; Hayward, N.K.; Montgomery, G.W.; Webb, P.M.; Whiteman, D.C. Single nucleotide polymorphisms in obesity related genes and the risk of esophageal cancers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Campa, D.; Pardini, B.; Naccarati, A.; Vodickova, L.; Novotny, J.; Steinke, V.; Rahner, N.; Holinski-Feder, E.; Morak, M.; Schackert, H.K.; et al. Polymorphisms of genes coding for ghrelin and its receptor in relation to colorectal cancer risk: A two-step gene-wide case-control study. Gastroenterology 2010, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, A. Role of the Ghrelin System in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 5380. [Google Scholar] [CrossRef]
- Dossus, L.; McKay, J.D.; Canzian, F.; Wilkening, S.; Rinaldi, S.; Biessy, C.; Olsen, A.; Tjønneland, A.; Jakobsen, M.U.; Overvad, K.; et al. Polymorphisms of genes coding for ghrelin and its receptor in relation to anthropometry, circulating levels of IGF-I and IGFBP-3, and breast cancer risk: A case–control study nested within the European Prospective Investigation into Cancer and Nutrition (EPIC). Carcinogenesis 2008, 29, 1360–1366. [Google Scholar] [CrossRef]
- Skibola, D.R.; Smith, M.T.; Bracci, P.M.; Hubbard, A.E.; Agana, L.; Chi, S.; Holly, E.A. Polymorphisms in ghrelin and neuropeptide Y genes are associated with non-Hodgkin lymphoma. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1251–1256. [Google Scholar] [CrossRef]
- Chopin, L.K.; Seim, I.; Walpole, C.M.; Herington, A.C. The ghrelin axis: Does it have an appetite for cancer progression? Endocr. Rev. 2012, 33, 849–891. [Google Scholar] [CrossRef]
- Han, Q.; Shan, Z.; Hu, J.; Zhang, N. Relationship between gene polymorphisms and prostate cancer risk. Asian Pac. J. Trop. Med. 2015, 8, 569–573. [Google Scholar] [CrossRef]
- Hormaechea-Agulla, D.; Gahete, M.D.; Jiménez-Vacas, J.M.; Gómez-Gómez, E.; Ibáñez-Costa, A.; López, F.L.; Rivero-Cortés, E.; Sarmento-Cabral, A.; Valero-Rosa, J.; Carrasco-Valiente, J.; et al. The oncogenic role of the In1-ghrelin splicing variant in prostate cancer aggressiveness. Mol. Cancer 2017, 16, 146. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vacas, J.M.; Montero-Hidalgo, A.J.; Gómez-Gómez, E.; Fuentes-Fayos, A.C.; Ruiz-Pino, F.; Guler, I.; Camargo, A.; Anglada, F.J.; Carrasco-Valiente, J.; Tena-Sempere, M.; et al. In1-Ghrelin Splicing Variant as a Key Element in the Pathophysiological Association Between Obesity and Prostate Cancer. J. Clin. Endocrinol. Metab. 2021, 106, e4956–e4968. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 15, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-Dimensionality Reduction Reveals High-Order Interactions among Estrogen Metabolism Genes in Sporadic Breast Cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Hahn, L.W.; Ritchie, M.D.; Moore, J.H. Multifactor dimensionality reduction software for detecting gene-gene and geneenvironment interactions. Bioinformatics 2003, 19, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Gilbert, J.C.; Tsai, C.T.; Chiang, F.T.; Holden, T.B.N.; White, B.C. A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J. Theor. Biol. 2006, 241, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Freeman, V.L.; Meydani, M.; Hurr, K.; Flanigan, R.C. Inverse association between prostatic polyunsaturated fatty acid and risk of locally advanced prostate carcinoma. Cancer 2004, 101, 2744–2754. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Diabetes, cancer, and metformin: Connections of metabolism and cell proliferation. Ann. N. Y. Acad. Sci. 2011, 1243, 54–68. [Google Scholar] [CrossRef]
- Travis, R.C.; Appleby, P.N.; Martin, R.M.; Holly, J.M.; Albanes, D.; Black, A.; Bueno-de-Mesquita, H.B.; Chan, J.M.; Chen, C.; Chirlaque, M.D.; et al. A meta-analysis of individual participant data reveals an association between circulating levels of IGF-I and prostate cancer risk. Cancer Res. 2016, 76, 2288–2300. [Google Scholar] [CrossRef]
- Zhang, X.; Zhai, L.; Rong, C.; Qin, X.; Shan, L. Association of Ghrelin Gene Polymorphisms and Serum Ghrelin Levels with the Risk of Hepatitis B Virus-Related Liver Diseases in a Chinese Population. PLoS ONE 2005, 10, e0143069. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, Z.X.; Guo, D.Y.; Tao, Y.X. Regulation of prostate cancer by hormone-responsive G protein-coupled receptors. Pharmacol. Ther. 2018, 191, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Huang, W.D.; Lv, X.Y.; Yang, Y.M. Ghrelin protects H9c2 cells from hydrogen peroxide-induced apoptosis through NF-kappaB and mitochondria-mediated signaling. Eur. J. Pharmacol. 2011, 654, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Garcia, E.A.; Korbonits, M. Genetic studies on the ghrelin, growth hormone secretagogue receptor (GHSR) and ghrelin O-acyl transferase (GOAT) genes. Peptides 2011, 32, 2191–2207. [Google Scholar] [CrossRef]
- Marzolini, C.; Paus, E.; Buclin, T.; Kim, R.B. Polymorphisms in human MDR1 (Pglycoprotein): Recent advances and clinical relevance. Clin. Pharmacol. Ther. 2004, 75, 13–33. [Google Scholar] [CrossRef]
- Espigolan, R.; Baldi, F.; Boligon, A.A.; Souza, F.R.; Gordo, D.G.; Tonussi, R.L.; Cardoso, D.F.; Oliveira, H.N.; Tonhati, H.; Sargolzaei, M.; et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genom. 2013, 14, 305. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Kijas, J.W.; Reverter, A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet. Sel. Evol. 2014, 46, 22. [Google Scholar] [CrossRef]
- Fleck, J.L.; Pavel, A.B.; Cassandras, C.G. Integrating mutation and gene expression cross-sectional data to infer cancer progression. BMC Syst. Biol. 2016, 10, 12. [Google Scholar] [CrossRef]
- Jia, P.; Zhao, Z. Impacts of somatic mutations on gene expression: An association perspective. Brief. Bioinform. 2017, 18, 413–425. [Google Scholar] [CrossRef]
- Lin, T.C.; Hsiao, M. Ghrelin and cancer progression. Biochim. Biophys. Acta BBA Rev. Cancer 2017, 1868, 51–57. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Guo, Y.; Murtaza, M.; Joseph, T.; Glessner, J.T.; Bailey, S.D.; Onland-Moret, N.C.; Lettre, G.; Ongen, H.; Rajagopalan, R.; et al. Meta-analysis of Dense Genecentric Association Studies Reveals Common and Uncommon Variants Associated with Height. Am. J. Hum. Genet. 2011, 88, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Fernández Torres, J.; Martínez Nava, G.A.; Zamudio Cuevas, Y.; Martínez Flores, K.; Espinosa Morales, R. Epistasis between ADIPOQ rs1501299 and PON1 rs662 polymorphisms is potentially associated with the development of knee osteoarthritis. Mol. Biol. Rep. 2019, 46, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, R.; Priya, S. Genetic interactions effects for cancer disease identification using computational models: A review. Med. Biol. Eng. Comput. 2021, 59, 733–758. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Williams, S.M. New Strategies for Identifying Gene-gene Interactions in Hypertension. Ann. Med. 2002, 34, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.I. Epistasis and the Genetics of Human Diseases. Comptes Rendus Biol. 2002, 328, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Kardia, S.L.R. Context-Dependent Genetic Effects in Hypertension. Curr. Hypertens. Rep. 2000, 2, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R. Epistasis and Complex Traits. In Epistasis and the Evolutionary Process; Wade, M., Brodie, B., III, Wolf, J., Eds.; Oxford University Press: New York, NY, USA, 2000; pp. 41–57. [Google Scholar]
- Herbert, A.; Gerry, N.P.; McQueen, M.B.; Heid, I.M.; Pfeufer, A.; Illig, T.; Wichmann, H.E.; Meitinger, T.; Hunter, D.; Hu, F.B.; et al. A Common Genetic Variant is Associated with Adult and Childhood Obesity. J. Comput. Biol. 2006, 312, 279–384. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at CLU and CR1 Associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Marchini, J.; Donnelley, P.; Cardon, L.R. Genome-Wide Strategies for Detecting Multiple Loci that Influence Complex Diseases. Nat. Genet. 2005, 37, 413–417. [Google Scholar] [CrossRef]
- Velez, D.R.; White, B.C.; Motsinger, A.A.; Bush, W.S.; Ritchie, M.D.; Williams, S.M.; Moore, J.H. A Balanced Accuracy Function for Epistasis Modeling in Imbalanced Dataset using Multifactor Dimensionality Reduction. Genet. Epidemiol. 2007, 31, 306–315. [Google Scholar] [CrossRef]
- Jiang, X.; Barmada, M.; Visweswaran, S. Identifying Genetic Interactions in Genome-Wide Data Using Bayesian Networks. Genet. Epidemiol. 2010, 34, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.H.; Williams, S.M. Epistasis and its implications for personal genetics. Am. J. Hum. Genet. 2009, 85, 309–320. [Google Scholar] [CrossRef]
- Manuguerra, M.; Matullo, G.; Veglia, F.; Autrup, H.; Dunning, A.M.; Garte, S.; Gormally, E.; Malaveille, C.; Guarrera, S.; Polidoro, S.; et al. Multi-factor dimensionality reduction applied to a large prospective investigation on gene-gene and gene-environment interactions. Carcinogenesis 2007, 28, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.I.; Fagerholm, R.; Nevanlinna, H.; BenÍtez, J. The importance of replication in gene-gene interaction studies: Multifactor dimensionality reduction applied to a two-stage breast cancer case-control study. Carcinogenesis 2008, 29, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Sajdak, S.; Pawlik, P.; Lianeri, M.; Jagodzinski, P.P. DNMT1, DNMT3A and DNMT3B gene variants in relation to ovarian cancer risk in the Polish population. Mol. Biol. Rep. 2013, 40, 4893–4899. [Google Scholar] [CrossRef]
- Marcus, M.W.; Raji, O.Y.; Duffy, S.W.; Young, R.P.; Hopkins, R.J.; Field, J.K. Incorporating epistasis interaction of genetic susceptibility single nucleotide polymorphisms in a lung cancer risk prediction model. Int. J. Oncol. 2016, 49, 361–370. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Characteristics | Controls | Patients | p-Value |
|---|---|---|---|
| Age (years) | |||
| Mean ± SD | 71.59 ± 8.476 | 74.63 ± 9.50 | 0.015 |
| BMI (kg/m2) | |||
| Mean ± SD | 22.89 ± 1.304 | 22.29 ± 2.034 | 0.013 |
| Residence | |||
| Urban | 76 | 81 | |
| Rural | 19 | 14 | |
| PSA (ng/mL) | |||
| Mean ± SD | 4.26 ± 3.48 | 80.72 ± 24.34 | 0.001 |
| Gleason Score | |||
| <7 (Low) | - | 38 (31.66) | |
| 7 (Intermediate) | - | 48 (40.00) | NA |
| >7 (High) | - | 34 (28.33) | |
| Clinical Stade | |||
| Localised | - | 70 (58.33) | |
| Advanced | - | 34 (28.33) | NA |
| Metastatic | - | 16 (13.33) | |
| Death | - | 19 (15.43) | NA |
| Gene | SNP | Position | Allele | MAF | Localization | SNP Type | TaqMan SNP |
|---|---|---|---|---|---|---|---|
| GHRL 3p25.3 | rs4684677 | 10286769 | T > A | 0.060 | Exon 4 | Missense (Gln 90 Leu) | C__25607748_10 |
| rs696217 | 10289773 | G > T | 0.077 | Exon 3 | Missense (Leu 72 Met) | C___3151003_20 | |
| rs34911341 | 10289835 | C > T | 0.007 | Exon 3 | Missense (Arg 51 Gln) | C__25607739_20 | |
| GHSR 3q26.31 | rs2948694 | 172447373 | T/C, G, T | 0.110 | Intron 1 | Intron | C__16174361_10 |
| rs572169 | 172447937 | C > T | 0.295 | Exon 1 | Silent | C___1079489_20 | |
| rs2922126 | 172449471 | T > A | 0.303 | 2KB upstream variant | 5′ flanking region/promoter | C___3261006_10 |
| Gene | SNPs | HWE | No-HWE | χ2 | MAF |
|---|---|---|---|---|---|
| GHRL | rs696217 | - | p = 2 × 10−4 | 8.021 | 0.199 |
| rs4684677 | p = 1.000 | - | 0.002 | 0.005 | |
| rs34911341 | p = 1.000 | - | - | 0.000 | |
| GHSR | rs2922126 | - | p = 5.106 × 10−4 | 15.336 | 0.353 |
| rs572169 | - | p = 2.876 × 10−7 | 17.941 | 0.275 | |
| rs2948694 | p = 1.000 | - | 0.183 | 0.032 |
| Gene | Locus | Allele | Controls | Patients | OR (95% CI) | p-Value |
|---|---|---|---|---|---|---|
| GHRL | rs34911341 | |||||
| C > T | C | 170 (1.000) | 200 (1.000) | - | - | |
| rs696217 | n = 168 | n = 214 | ||||
| G > T | G | 138 (0.821) | 168 (0.785) | 0.794 (0.475~1.325) | 0.376 | |
| T | 30 (0.178) | 46 (0.215) | ||||
| rs4684677 | n = 172 | n = 208 | ||||
| A > G | A | 171 (0.994) | 207 (0.995) | 1.210 (0.075~19.497) | 0.892 | |
| G | 1 (0.005) | 1 (0.004) | ||||
| GHSR | ||||||
| rs2922126 | n = 174 | n = 240 | ||||
| T > A | A | 118 (0.678) | 150 (0.625) | 0.791 (0.524~1.193) | 0.263 | |
| T | 56 (0.321) | 90 (0.375) | ||||
| rs572169 | n = 160 | n = 204 | ||||
| C > T | C | 118 (0.738) | 147 (0.721) | 0.918 (0.575~1.463) | 0.718 | |
| T | 42 (0.263) | 57 (0.263) | ||||
| rs2948694 | n = 170 | n = 238 | ||||
| T/C, G, A | A | 167 (0.982) | 229 (0.962) | 2.188 (0.121~1.714) | 0.234 | |
| G | 3 (0.017) | 9 (0.037) |
| Gene | Locus | Genotype | Controls | Patients | OR (95% CI) | p-Value |
|---|---|---|---|---|---|---|
| GHRL | rs34911341 | C/C | 85 (1.000) | 100 (1.000) | - | - |
| rs696217 | G/G | 54 (0.642) | 61 (0.570) | 0.736 (0.409–1.326) | 0.307 | |
| G/T | 30 (0.357) | 46 (0.429) | ||||
| rs4684677 | A/A | 85 (0.988) | 103 (0.990) | 1.211 (0.074~19.663) | 0.892 | |
| A/G | 1 (0.011) | 1 (0.009) | ||||
| GHSR | rs2922126 | A/A | 31 (0.356) | 36 (0.300) | 1.292 (0.714–2.317) | 0.452 |
| A/T | 56 (0.644) | 78 (0.650) | 0.972 (0.555–1.729) | 0.999 | ||
| T/T | 0 (0.000) | 6 (0.050) | - | 0.040 | ||
| rs572169 | C/C | 38 (0.475) | 45 (0.441) | 0.872 (0.484–1.570) | 0.649 | |
| C/T | 42 (0.525) | 57 (0.559) | ||||
| 82 (0.965) | ||||||
| rs2948694 | A/A | 3 (0.035) | 110 (0.924) | 0.447 (0.117–1.7703) | 0.227 | |
| A/G | 9 (0.076) |
| Gene | Haplotype # | Frequencies | Controls | Patients | χ2 | p-Value |
|---|---|---|---|---|---|---|
| GHRL | AGC | 0.798 | 0.815 | 0.784 | 0.574 | 0.448 |
| ATC | 0.197 | 0.179 | 0.211 | 0.621 | 0.430 | |
| GHSR | ACA | 0.587 | 0.599 | 0.577 | 0.195 | 0.658 |
| ATT | 0.211 | 0.191 | 0.226 | 0.747 | 0.387 | |
| ACT | 0.121 | 0.122 | 0.120 | 0.002 | 0.968 | |
| ATA | 0.048 | 0.065 | 0.036 | 1.757 | 0.184 | |
| GTA | 0.018 | 0.016 | 0.019 | 0.061 | 0.805 | |
| GCT | 0.015 | 0.007 | 0.020 | 1.156 | 0.282 |
| Modèles | Training Balanced Accuracy | Testing Balanced Accuracy | CVC | F-Value | MCC |
|---|---|---|---|---|---|
| rs2922126 | 0.6780 | 0.5631 | 7/10 | - | - |
| rs572169, rs2922126 | 0.8235 | 0.8108 | 10/10 | 0.953 | 1.544 |
| rs572169, rs696217, rs2922126 | 0.8335 | 0.8198 | 7/10 | - | - |
| rs572169, rs34911341, rs696217, rs2922126 | 0.8370 | 0.8198 | 7/10 | - | - |
| rs572169, rs34911341, rs696217, rs4684677, rs2922126 | 0.8003 | 0.6622 | 10/10 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merabet, N.; Ramoz, N.; Boulmaiz, A.; Bourefis, A.; Benabdelkrim, M.; Djeffal, O.; Moyse, E.; Tolle, V.; Berredjem, H. SNPs-Panel Polymorphism Variations in GHRL and GHSR Genes Are Not Associated with Prostate Cancer. Biomedicines 2023, 11, 3276. https://doi.org/10.3390/biomedicines11123276
Merabet N, Ramoz N, Boulmaiz A, Bourefis A, Benabdelkrim M, Djeffal O, Moyse E, Tolle V, Berredjem H. SNPs-Panel Polymorphism Variations in GHRL and GHSR Genes Are Not Associated with Prostate Cancer. Biomedicines. 2023; 11(12):3276. https://doi.org/10.3390/biomedicines11123276
Chicago/Turabian StyleMerabet, Nesrine, Nicolas Ramoz, Amel Boulmaiz, Asma Bourefis, Maroua Benabdelkrim, Omar Djeffal, Emmanuel Moyse, Virginie Tolle, and Hajira Berredjem. 2023. "SNPs-Panel Polymorphism Variations in GHRL and GHSR Genes Are Not Associated with Prostate Cancer" Biomedicines 11, no. 12: 3276. https://doi.org/10.3390/biomedicines11123276