
Thrombo-Inflammation in COVID-19 and Sickle Cell Disease: Two Faces of the Same Coin


Abstract
:1. Introduction
2. Pathophysiology of Sickle Cell Disease and COVID-19
3. Mechanisms of Multicellular Adhesion and Thrombo-Inflammation in Sickle Cell Disease and COVID-19
3.1. Endothelial Cell Injury and Activation: Role in Thrombo-Inflammation in SCD and COVID-19
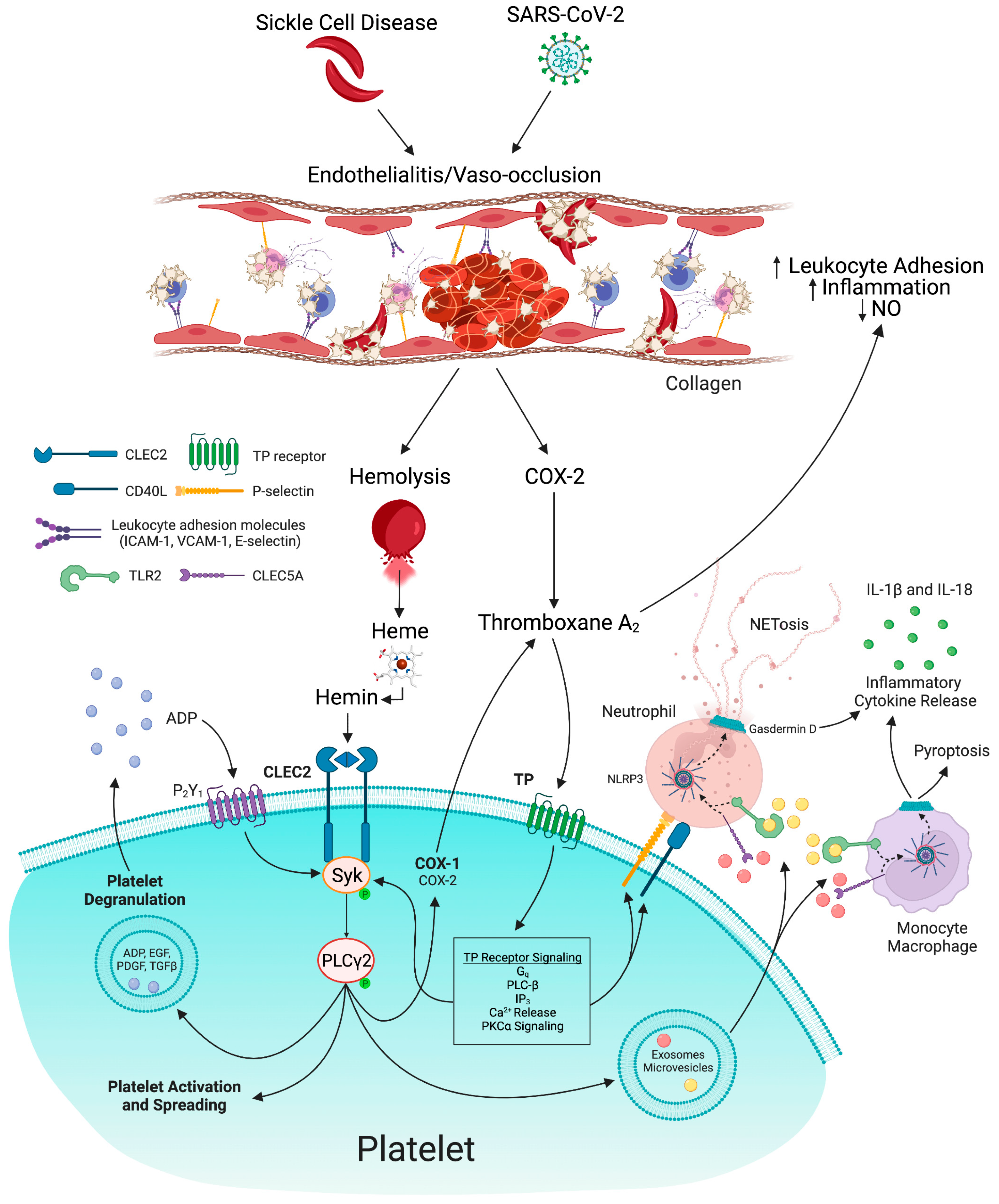
3.2. Platelet Activation: Role in Thrombo-Inflammation in SCD and COVID-19
3.3. P-Selectin: Role in Thrombo-Inflammation in SCD and COVID-19
3.4. Tissue Factor: Role in Thrombo-Inflammation in SCD and COVID-19
3.5. CD40L: Role in Thrombo-Inflammation in SCD and COVID-19
3.6. NLRP3 Inflammasome: Role in Thromboinflammation in SCD and COVID-19
3.7. Nitric Oxide: Role in Thrombo-Inflammation in SCD and COVID-19
3.8. TGFβ: Role in Thromboinflammation in SCD and COVID-19
3.9. Lipoxygenase: Role in Thrombo-Inflammation in SCD and COVID-19
4. Thromboxane A2-A Key Mediator of Thrombo-inflammation by Regulation of Platelet Activation, NO Synthesis, and Expression of P-Selectin, CD40L, Tissue Factor, and TGF-β
4.1. Thromboxane A2-Mediated P-Selectin Expression
4.2. Thromboxane A2-Mediated Tissue Factor Expression
4.3. Thromboxane A2-Mediated CD40L Expression
4.4. Thromboxane A2-Induced Suppression of NO Synthesis
4.5. Thromboxane A2-Induced TGF-β Release
5. Thromboxane A2 in Post-Capillary Venoconstriction in SCD and COVID-19
5.1. Post-Capillary Pulmonary Venous Constriction
5.2. Post-Capillary Efferent Arteriole Constriction in Kidney Injury
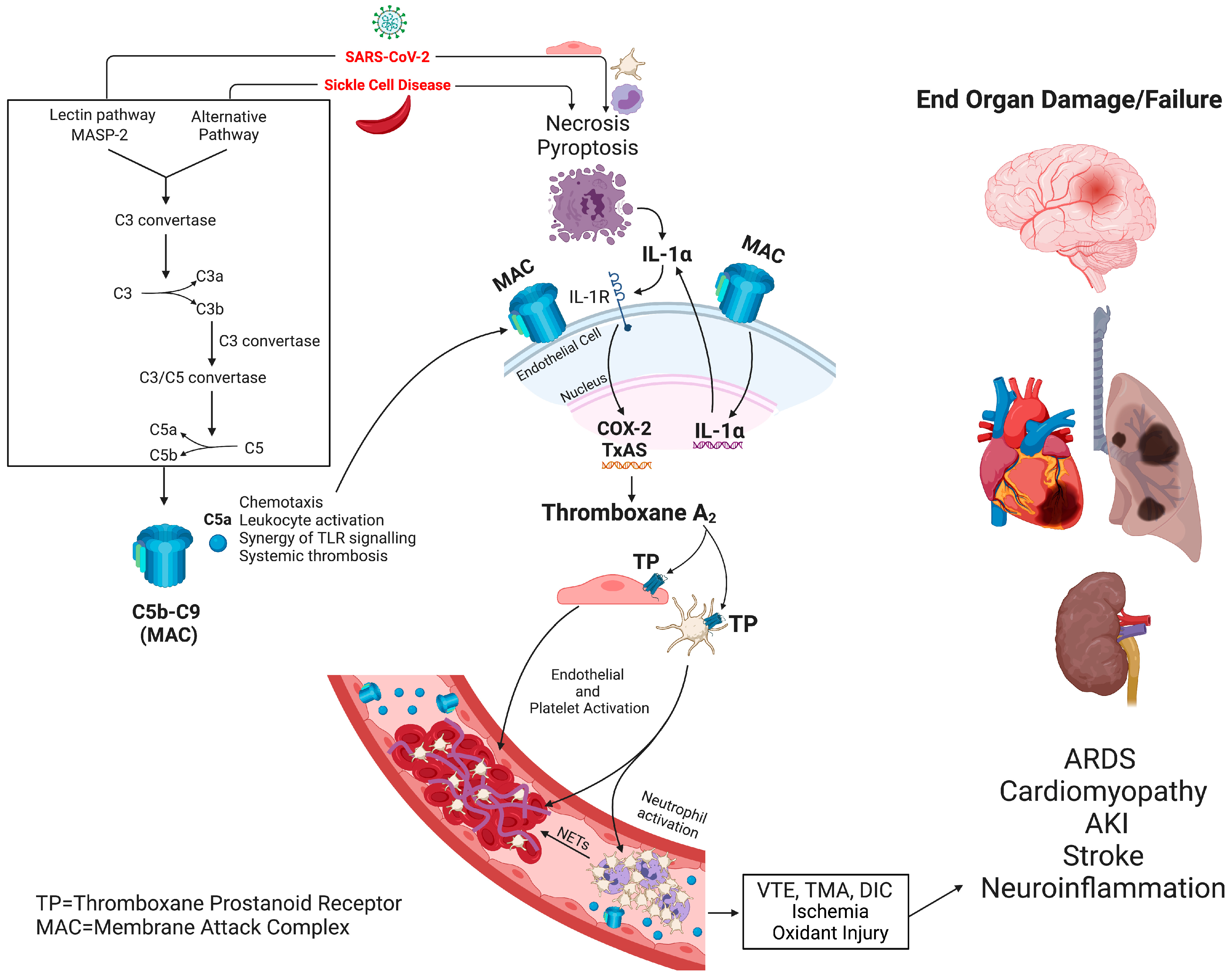
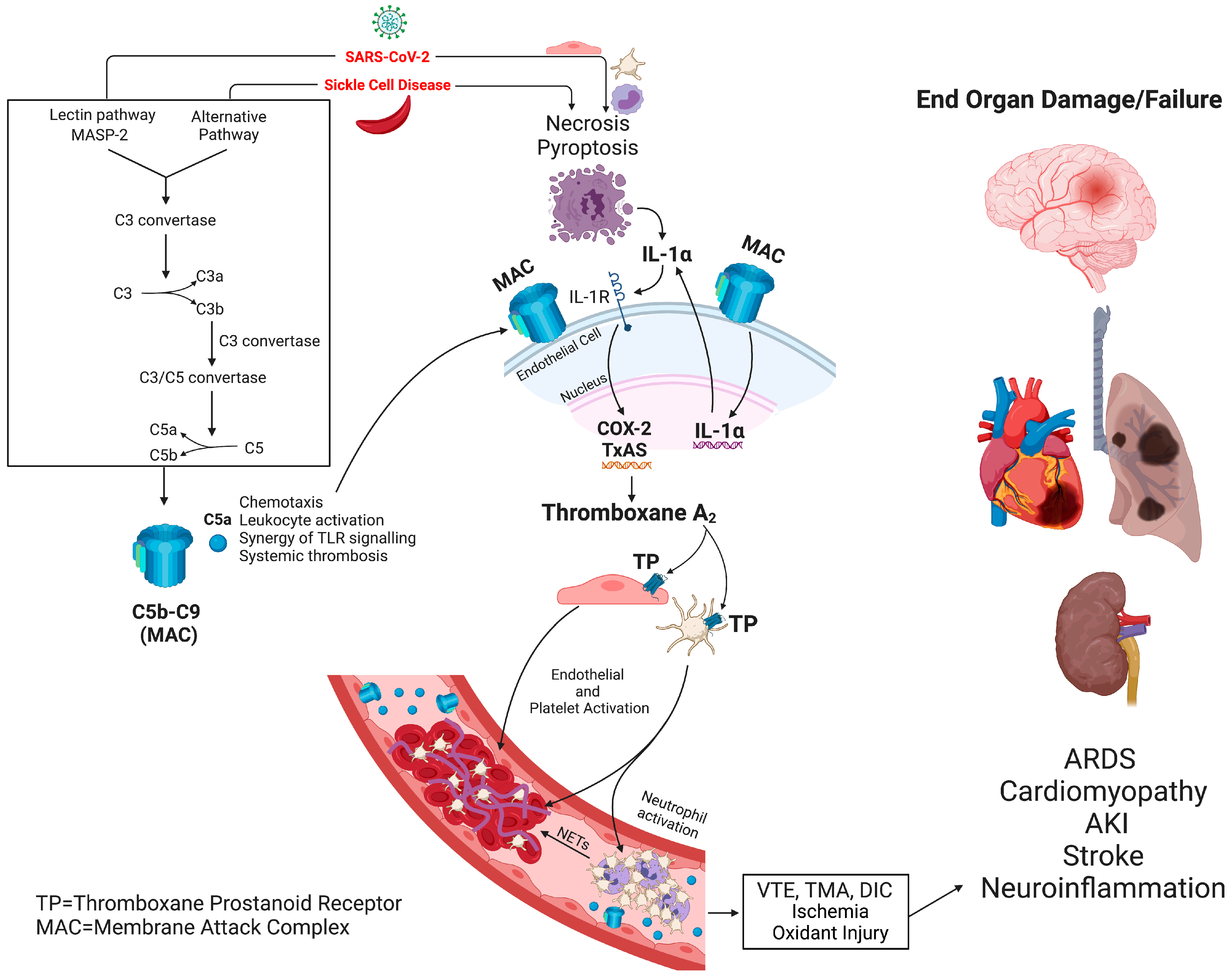
6. Complement Activation as an Inducer of Thrombo-Inflammation in SCD and COVID-19
7. Thromboxane A2 Is Enzymatically Converted into 11-Dehydro-Thromboxane A2, a Full Agonist of the Prostaglandin D2/DP2 Receptor Leading to Fibrosis and Inflammation
8. Therapeutic Options for Thrombo-inflammation in COVID-19 and SCD: Past, Present, and Future
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Singh, A.; Brandow, A.M.; Panepinto, J.A. COVID-19 in individuals with sickle cell disease/trait compared with other Black individuals. Blood Adv. 2021, 5, 1915–1921. [Google Scholar] [CrossRef]
- Panepinto, J.A.; Brandow, A.; Mucalo, L.; Yusuf, F.; Singh, A.; Taylor, B.; Woods, K.; Payne, A.B.; Peacock, G.; Schieve, L.A. Coronavirus Disease among Persons with Sickle Cell Disease, United States, March 20–May 21, 2020. Emerg. Infect. Dis. 2020, 26, 2473–2476. [Google Scholar] [CrossRef]
- Minniti, C.P.; Zaidi, A.U.; Nouraie, M.; Manwani, D.; Crouch, G.D.; Crouch, A.S.; Callaghan, M.U.; Carpenter, S.; Jacobs, C.; Han, J.; et al. Clinical predictors of poor outcomes in patients with sickle cell disease and COVID-19 infection. Blood Adv. 2021, 5, 207–215. [Google Scholar] [CrossRef]
- Boga, C.; Asma, S.; Leblebisatan, G.; Sen, N.; Tombak, A.; Demiroglu, Y.Z.; Yeral, M.; Akin, S.; Yesilagac, H.; Habesoglu, M.A.; et al. Comparison of the clinical course of COVID-19 infection in sickle cell disease patients with healthcare professionals. Ann. Hematol. 2021, 100, 2195–2202. [Google Scholar] [CrossRef]
- Arlet, J.B.; de Luna, G.; Khimoud, D.; Odievre, M.H.; de Montalembert, M.; Joseph, L.; Chantalat-Auger, C.; Flamarion, E.; Bartolucci, P.; Lionnet, F.; et al. Prognosis of patients with sickle cell disease and COVID-19: A French experience. Lancet Haematol. 2020, 7, e632–e634. [Google Scholar] [CrossRef]
- Alkindi, S.; Elsadek, R.A.; Al-Madhani, A.; Al-Musalhi, M.; AlKindi, S.Y.; Al-Khadouri, G.; Al Rawahi, B.; Al-Ruqeishi, S.; Al-Yazeedi, J.; Wali, Y.A.; et al. Impact of COVID-19 on vasooclusive crisis in patients with sickle cell anaemia. Int. J. Infect. Dis. 2021, 106, 128–133. [Google Scholar] [CrossRef]
- Brousse, V.; Holvoet, L.; Pescarmona, R.; Viel, S.; Perret, M.; Visseaux, B.; Ferre, V.M.; Ithier, G.; Le Van Kim, C.; Benkerrou, M.; et al. Low incidence of COVID-19 severe complications in a large cohort of children with sickle cell disease: A protective role for basal interferon-1 activation? Haematologica 2021, 106, 2746–2748. [Google Scholar] [CrossRef]
- Patricia, H.; Slim, A.; Emilie-Fleur, G.; François, G.; Vincent, B.; Darragh, D.; Sebastien, D.; Pierre-Louis, T.; Patrick, M.; Caroline Le Van, K.; et al. The proteome of neutrophils in sickle cell disease reveals an unexpected activation of interferon alpha signaling pathway. Haematologica 2020, 105, 2851–2854. [Google Scholar] [CrossRef] [Green Version]
- Madany, E.; Okwan-Duodu, D.; Balbuena-Merle, R.; Hendrickson, J.E.; Gibb, D.R. Potential Implications of a Type 1 Interferon Gene Signature on COVID-19 Severity and Chronic Inflammation in Sickle Cell Disease. Front. Med. 2021, 8, 679030. [Google Scholar] [CrossRef]
- Verma, A.; Huffman, J.E.; Gao, L.; Minnier, J.; Wu, W.-C.; Cho, K.; Ho, Y.-L.; Gorman, B.R.; Pyarajan, S.; Rajeevan, N.; et al. Association of Kidney Comorbidities and Acute Kidney Failure with Unfavorable Outcomes after COVID-19 in Individuals with the Sickle Cell Trait. JAMA Intern. Med. 2022, 182, 796–804. [Google Scholar] [CrossRef]
- Dun, C.; Walsh, C.M.; Bae, S.; Adalja, A.; Toner, E.; Lash, T.A.; Hashim, F.; Paturzo, J.; Segev, D.L.; Makary, M.A. A Machine Learning Study of 534,023 Medicare Beneficiaries with COVID-19: Implications for Personalized Risk Prediction. medRxiv 2020. [Google Scholar] [CrossRef]
- CDC. Data and Statistics on Sickle Cell Disease. Available online: https://www.cdc.gov/ncbddd/sicklecell/data.html (accessed on 20 February 2022).
- American Society of Hematology. Rare Patients with Sickle Cell Disease Live Nearly Twice as Long as Average. Available online: https://www.hematology.org/newsroom/press-releases/2016/rare-patients-with-sickle-cell-disease-live-nearly-twice-as-long-as-average# (accessed on 20 February 2022).
- Adams, R.J.; McKie, V.C.; Hsu, L.; Files, B.; Vichinsky, E.; Pegelow, C.; Abboud, M.; Gallagher, D.; Kutlar, A.; Nichols, F.T.; et al. Prevention of a First Stroke by Transfusions in Children with Sickle Cell Anemia and Abnormal Results on Transcranial Doppler Ultrasonography. N. Engl. J. Med. 1998, 339, 5–11. [Google Scholar] [CrossRef]
- Arun, S.S.; Maria, A.L.-I.; Rakhi, P.N. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica 2020, 105, 2368–2379. [Google Scholar] [CrossRef]
- Usmani, A.; Machado, R.F. Vascular complications of sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 205–221. [Google Scholar] [CrossRef]
- Cushman, M.; Cantrell, R.A.; McClure, L.A.; Howard, G.; Prineas, R.J.; Moy, C.S.; Temple, E.M.; Howard, V.J. Estimated 10-year stroke risk by region and race in the United States: Geographic and racial differences in stroke risk. Ann. Neurol. 2008, 64, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Naik, R.P.; Streiff, M.B.; Haywood, C.; Segal, J.B.; Lanzkron, S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J. Thromb. Haemost. 2014, 12, 2010–2016. [Google Scholar] [CrossRef] [Green Version]
- Brunson, A.; Lei, A.; Rosenberg, A.S.; White, R.H.; Keegan, T.; Wun, T. Increased incidence of VTE in sickle cell disease patients: Risk factors, recurrence and impact on mortality. Br. J. Haematol. 2017, 178, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R., Jr.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of COVID-19 in New York City. N. Engl. J. Med. 2020, 382, 2372–2374. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregates formation trigger tissue factor expression in severe COVID-19 patients. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-C.; Fitzgerald, G.A. COVID-19, microangiopathy, hemostatic activation, and complement. J. Clin. Investig. 2020, 130, 3950–3953. [Google Scholar] [CrossRef] [PubMed]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Vats, R.; Brzoska, T.; Bennewitz, M.F.; Jimenez, M.A.; Pradhan-Sundd, T.; Tutuncuoglu, E.; Jonassaint, J.; Gutierrez, E.; Watkins, S.C.; Shiva, S.; et al. Platelet Extracellular Vesicles Drive Inflammasome-IL-1β-Dependent Lung Injury in Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2020, 201, 33–46. [Google Scholar] [CrossRef]
- John, N.A.; John, J.; Kamblec, P.; Singhal, A.; Daulatabad, V.; Vamshidhar, I.S. Patients with Sticky Platelet Syndrome, Sickle Cell Disease and Glanzmann Syndrome May Promulgate Severe Thrombosis if Infected with COVID-19. Maedica 2021, 16, 268–273. [Google Scholar] [CrossRef]
- Jain, S.; Bakshi, N.; Krishnamurti, L. Acute Chest Syndrome in Children with Sickle Cell Disease. Pediatr. Allergy Immunol. Pulmonol. 2017, 30, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Castro, O.; Brambilla, D.J.; Thorington, B.; Reindorf, C.A.; Scott, R.B.; Gillette, P.; Vera, J.C.; Levy, P.S. The acute chest syndrome in sickle cell disease: Incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994, 84, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, S.R. Management of respiratory failure due to COVID-19. BMJ 2020, 369, m1786. [Google Scholar] [CrossRef]
- Mekontso Dessap, A.; Deux, J.F.; Habibi, A.; Abidi, N.; Godeau, B.; Adnot, S.; Brun-Buisson, C.; Rahmouni, A.; Galacteros, F.; Maitre, B. Lung imaging during acute chest syndrome in sickle cell disease: Computed tomography patterns and diagnostic accuracy of bedside chest radiograph. Thorax 2014, 69, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Dako, F.; Hossain, R.; Jeudy, J.; White, C. Dual-energy CT evidence of pulmonary microvascular occlusion in patients with sickle cell disease experiencing acute chest syndrome. Clin. Imaging 2021, 78, 94–97. [Google Scholar] [CrossRef]
- Cao, Y.; Geng, C.; Li, Y.; Zhang, Y. In situ Pulmonary Artery Thrombosis: A Previously Overlooked Disease. Front. Pharmacol. 2021, 12, 671589. [Google Scholar] [CrossRef]
- Nouraie, M.; Zhang, X.; Srisuwananukorn, A.; Machado, R.F.; Gordeuk, V.R.; Gladwin, M.T.; Saraf, S. Potential Contribution of Pulmonary Thromboembolic Disease in Pulmonary Hypertension in Sickle Cell Disease. Ann. Am. Thorac. Soc. 2020, 17, 899–901. [Google Scholar] [CrossRef]
- Winchenne, A.; Cecchini, J.; Deux, J.F.; De Prost, N.; Razazi, K.; Carteaux, G.; Galacteros, F.; Habibi, A.; Bartolucci, P.; Melica, G.; et al. A clinical risk score for pulmonary artery thrombosis during acute chest syndrome in adult patients with sickle cell disease. Br. J. Haematol. 2017, 179, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Adedeji, M.O.; Cespedes, J.; Allen, K.; Subramony, C.; Hughson, M.D. Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Arch. Pathol. Lab. Med. 2001, 125, 1436–1441. [Google Scholar] [CrossRef]
- Thomas, A.N.; Pattison, C.; Serjeant, G.R. Causes of death in sickle-cell disease in Jamaica. Br. Med. J. Clin. Res. Ed. 1982, 285, 633–635. [Google Scholar] [CrossRef] [Green Version]
- Mekontso Dessap, A.; Deux, J.F.; Abidi, N.; Lavenu-Bombled, C.; Melica, G.; Renaud, B.; Godeau, B.; Adnot, S.; Brochard, L.; Brun-Buisson, C.; et al. Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1022–1029. [Google Scholar] [CrossRef]
- Vichinsky, E.P.; Neumayr, L.D.; Earles, A.N.; Williams, R.; Lennette, E.T.; Dean, D.; Nickerson, B.; Orringer, E.; McKie, V.; Bellevue, R.; et al. Causes and Outcomes of the Acute Chest Syndrome in Sickle Cell Disease. N. Engl. J. Med. 2000, 342, 1855–1865. [Google Scholar] [CrossRef] [Green Version]
- Vichinsky, E.; Williams, R.; Das, M.; Earles, A.N.; Lewis, N.; Adler, A.; McQuitty, J. Pulmonary fat embolism: A distinct cause of severe acute chest syndrome in sickle cell anemia. Blood 1994, 83, 3107–3112. [Google Scholar] [CrossRef] [Green Version]
- Dang, N.C.; Johnson, C.; Eslami-Farsani, M.; Haywood, L.J. Bone marrow embolism in sickle cell disease: A review. Am. J. Hematol. 2005, 79, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Godeau, B.; Schaeffer, A.; Bachir, D.; Fleury-Feith, J.; Galacteros, F.; Verra, F.; Escudier, E.; Vaillant, J.N.; Brun-Buisson, C.; Rahmouni, A.; et al. Bronchoalveolar lavage in adult sickle cell patients with acute chest syndrome: Value for diagnostic assessment of fat embolism. Am. J. Respir. Crit. Care Med. 1996, 153, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Lechapt, E.; Habibi, A.; Bachir, D.; Galacteros, F.; Schaeffer, A.; Desvaux, D.; Brochard, L.; Housset, B.; Godeau, B.; Maitre, B. Induced sputum versus bronchoalveolar lavage during acute chest syndrome in sickle cell disease. Am. J. Respir. Crit. Care Med. 2003, 168, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Anea, C.B.; Lyon, M.; Lee, I.A.; Gonzales, J.N.; Adeyemi, A.; Falls, G.; Kutlar, A.; Brittain, J.E. Pulmonary platelet thrombi and vascular pathology in acute chest syndrome in patients with sickle cell disease. Am. J. Hematol. 2016, 91, 173–178. [Google Scholar] [CrossRef]
- Ballas, S. The Evolving Pharmacotherapeutic Landscape for the Treatment of Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020010. [Google Scholar] [CrossRef]
- Vichinsky, E. Emerging ‘A’ therapies in hemoglobinopathies: Agonists, antagonists, antioxidants, and arginine. Hematology 2012, 2012, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Kotiah, S.D.; Ballas, S.K. Investigational drugs in sickle cell anemia. Expert Opin. Investig. Drugs 2009, 18, 1817–1828. [Google Scholar] [CrossRef]
- Zennadi, R.; Moeller, B.J.; Whalen, E.J.; Batchvarova, M.; Xu, K.; Shan, S.; Delahunty, M.; Dewhirst, M.W.; Telen, M.J. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood 2007, 110, 2708–2717. [Google Scholar] [CrossRef]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Archambault, A.-S.; Zaid, Y.; Rakotoarivelo, V.; Doré, É.; Dubuc, I.; Martin, C.; Amar, Y.; Cheikh, A.; Fares, H.; El Hassani, A.; et al. Lipid storm within the lungs of severe COVID-19 patients: Extensive levels of cyclooxygenase and lipoxygenase-derived inflammatory metabolites. medRxiv 2020. [Google Scholar] [CrossRef]
- Solovey, A.; Lin, Y.; Browne, P.; Choong, S.; Wayner, E.; Hebbel, R.P. Circulating activated endothelial cells in sickle cell anemia. N. Engl. J. Med. 1997, 337, 1584–1590. [Google Scholar] [CrossRef]
- Carden, M.A.; Little, J. Emerging disease-modifying therapies for sickle cell disease. Haematologica 2019, 104, 1710–1719. [Google Scholar] [CrossRef] [Green Version]
- Shet, A.S.; Aras, O.; Gupta, K.; Hass, M.J.; Rausch, D.J.; Saba, N.; Koopmeiners, L.; Key, N.S.; Hebbel, R.P. Sickle blood contains tissue factor–positive microparticles derived from endothelial cells and monocytes. Blood 2003, 102, 2678–2683. [Google Scholar] [CrossRef] [Green Version]
- Solovey, A.; Gui, L.; Key, N.S.; Hebbel, R.P. Tissue factor expression by endothelial cells in sickle cell anemia. J. Clin. Investig. 1998, 101, 1899–1904. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Xu, C.; Manwani, D.; Frenette, P.S. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016, 127, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.S.; De Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Vats, R.; Kaminski, T.W.; Brzoska, T.; Leech, J.A.; Tutuncuoglu, E.; Katoch, O.; Jonassaint, J.C.; Tejero, J.; Novelli, E.M.; Pradhan-Sundd, T.; et al. Liver to lung microembolic NETs promote Gasdermin-D-dependent inflammatory lung injury in Sickle Cell Disease. Blood 2022, 140, 1020–1037. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Joshua, H.B.; Martina, C.; Ying, D.; Eleyna, M.; Alexander, S.; Lubka, T.R.; Jordan, D.D.; Steve, P.W.; Julie, R. Heme induces human and mouse platelet activation through C-type-lectin-like receptor-2. Haematologica 2020, 106, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, R.; Silveira, A.A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Pitanga, T.N.; Oliveira, R.R.; Zanette, D.L.; Guarda, C.C.; Santiago, R.P.; Santana, S.S.; Nascimento, V.M.L.; Lima, J.B.; Carvalho, G.Q.; Maffili, V.V.; et al. Sickle red cells as danger signals on proinflammatory gene expression, leukotriene B4 and interleukin-1 beta production in peripheral blood mononuclear cell. Cytokine 2016, 83, 75–84. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Kaul, D.K.; Finnegan, E.; Barabino, G.A. Sickle Red Cell–Endothelium Interactions. Microcirculation 2009, 16, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Scheim, D. From Cold to Killer: How SARS-CoV-2 Evolved without Hemagglutinin Esterase to Agglutinate, Then Clot Blood Cells in Pulmonary and Systemic Microvasculature. SSRN 2020, 28. [Google Scholar] [CrossRef]
- Modrof, J.; Kerschbaum, A.; Farcet, M.R.; Niemeyer, D.; Corman, V.M.; Kreil, T.R. SARS-CoV-2 and the safety margins of cell-based biological medicinal products. Biologicals 2020, 68, 122–124. [Google Scholar] [CrossRef]
- Lam, L.M.; Murphy, S.J.; Kuri-Cervantes, L.; Weisman, A.R.; Ittner, C.A.G.; Reilly, J.P.; Pampena, M.B.; Betts, M.R.; Wherry, E.J.; Song, W.-C.; et al. Erythrocytes Reveal Complement Activation in Patients with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Diorio, C.; McNerney, K.O.; Lambert, M.; Paessler, M.; Anderson, E.M.; Henrickson, S.E.; Chase, J.; Liebling, E.J.; Burudpakdee, C.; Lee, J.H.; et al. Evidence of thrombotic microangiopathy in children with SARS-CoV-2 across the spectrum of clinical presentations. Blood Adv. 2020, 4, 6051–6063. [Google Scholar] [CrossRef]
- Sweeney, J.M.; Barouqa, M.; Krause, G.J.; Gonzalez-Lugo, J.D.; Rahman, S.; Gil, M.R. Evidence for secondary thrombotic microangiopathy in COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- He, X.; Yao, F.; Chen, J.; Wang, Y.; Fang, X.; Lin, X.; Long, H.; Wang, Q.; Wu, Q. The poor prognosis and influencing factors of high D-dimer levels for COVID-19 patients. Sci. Rep. 2021, 11, 1830. [Google Scholar] [CrossRef]
- Dar, J.; Mughal, I.; Mekki, T.; Chapunduka, Z.; Hassan, I. Raised D-dimer levels in acute sickle cell crisis and their correlation with chest X-ray abnormalities. Ger. Med. Sci. GMS E J. 2010, 8, Doc25. [Google Scholar] [CrossRef]
- Francis, R.B., Jr. Elevated fibrin D-dimer fragment in sickle cell anemia: Evidence for activation of coagulation during the steady state as well as in painful crisis. Haemostasis 1989, 19, 105–111. [Google Scholar] [CrossRef]
- Bosevski, M.; Krstevski, G.; Bosevska, G.; Kapsarov, K.; Dodic, E.; Feehan, J.; Stojanoska, L.; Apostolopoulos, V. The role of D-dimer in relation to the clinical course of patients with COVID-19. Acta Biochim. Biophys. Sin. 2020, 53, 119–120. [Google Scholar] [CrossRef]
- Mouhat, B.; Besutti, M.; Bouiller, K.; Grillet, F.; Monnin, C.; Ecarnot, F.; Behr, J.; Capellier, G.; Soumagne, T.; Pili-Floury, S.; et al. Elevated D-dimers and lack of anticoagulation predict PE in severe COVID-19 patients. Eur. Respir. J. 2020, 56, 2001811. [Google Scholar] [CrossRef]
- Nebor, D.; Bowers, A.; Connes, P.; Hardy-Dessources, M.-D.; Knight-Madden, J.; Cumming, V.; Reid, M.; Romana, M. Plasma Concentration of Platelet-Derived Microparticles Is Related to Painful Vaso-Occlusive Phenotype Severity in Sickle Cell Anemia. PLoS ONE 2014, 9, e87243. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Haj, R.B.E.; et al. Platelets Can Associate with SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Theken, K.N.; Fitzgerald, G.A. Bioactive lipids in antiviral immunity. Science 2021, 371, 237–238. [Google Scholar] [CrossRef]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef]
- Sung, P.-S.; Huang, T.-F.; Hsieh, S.-L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badolia, R.; Inamdar, V.; Manne, B.K.; Dangelmaier, C.; Eble, J.A.; Kunapuli, S.P. G(q) pathway regulates proximal C-type lectin-like receptor-2 (CLEC-2) signaling in platelets. J. Biol. Chem. 2017, 292, 14516–14531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2016, 376, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Bennewitz, M.F.; Jimenez, M.A.; Vats, R.; Tutuncuoglu, E.; Jonassaint, J.; Kato, G.J.; Gladwin, M.T.; Sundd, P. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2017, 2, e89761. [Google Scholar] [CrossRef] [Green Version]
- Voskaridou, E.; Aimilia, M.; Flevari, P.; Dimopoulou, M.; Komninaka, V.; Repa, K.; Papassotiriou, I. Soluble P-Selectin Levels in Patients with Sickle Cell Disease Reflect Platelets’ Activation Rather Than Endothelial Dysfunction. Blood 2019, 134, 4829. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Leucker, T.M.; Osburn, W.O.; Reventun, P.; Smith, K.; Claggett, B.; Kirwan, B.-A.; de Brouwer, S.; Williams, M.S.; Gerstenblith, G.; Hager, D.N.; et al. Effect of Crizanlizumab, a P-Selectin Inhibitor, in COVID-19: A Placebo-Controlled, Randomized Trial. JACC Basic Transl. Sci. 2021, 6, 935–945. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. Role of Tissue Factor in Hemostasis, Thrombosis, and Vascular Development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Pawlinski, R.; Pedersen, B.; Erlich, J.; Mackman, N. Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: Lessons from low tissue factor mice. Thromb. Haemost. 2004, 92, 444–450. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chen, C.; Brzoska, T.; Nguyen, J.; Wang, S.; Vercellotti, G.M.; Key, N.S.; Sundd, P.; Belcher, J.D.; Pawlinski, R. Thrombin activation of PAR-1 contributes to microvascular stasis in mouse models of sickle cell disease. Blood 2020, 135, 1783–1787. [Google Scholar] [CrossRef]
- Karpusas, M.; Hsu, Y.M.; Wang, J.H.; Thompson, J.; Lederman, S.; Chess, L.; Thomas, D. 2 A crystal structure of an extracellular fragment of human CD40 ligand. Structure 1995, 3, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Elgueta, R.; Benson, M.J.; De Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.P.; Ataga, K.I.; Orringer, E.P.; Phillips, D.R.; Parise, L.V. Biologically active CD40 ligand is elevated in sickle cell anemia: Potential role for platelet-mediated inflammation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1626–1631. [Google Scholar] [CrossRef]
- Nicola, C.; Erich, V.D.P. Thromboinflammatory mechanisms in sickle cell disease—Challenging the hemostatic balance. Haematologica 2020, 105, 2380–2390. [Google Scholar] [CrossRef]
- Novelli, E.M.; Kato, G.J.; Ragni, M.V.; Zhang, Y.; Hildesheim, M.E.; Nouraie, M.; Barge, S.; Meyer, M.P.; Hassett, A.C.; Gordeuk, V.R.; et al. Plasma thrombospondin-1 is increased during acute sickle cell vaso-occlusive events and associated with acute chest syndrome, hydroxyurea therapy, and lower hemolytic rates. Am. J. Hematol. 2012, 87, 326–330. [Google Scholar] [CrossRef]
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 70–78. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Courjon, J.; Dufies, O.; Robert, A.; Bailly, L.; Torre, C.; Chirio, D.; Contenti, J.; Vitale, S.; Loubatier, C.; Doye, A.; et al. Heterogeneous NLRP3 inflammasome signature in circulating myeloid cells as a biomarker of COVID-19 severity. Blood Adv. 2021, 5, 1523–1534. [Google Scholar] [CrossRef]
- Rolfes, V.; Ribeiro, L.S.; Hawwari, I.; Bottcher, L.; Rosero, N.; Maasewerd, S.; Santos, M.L.S.; Prochnicki, T.; Silva, C.M.S.; Wanderley, C.W.S.; et al. Platelets Fuel the Inflammasome Activation of Innate Immune Cells. Cell Rep. 2020, 31, 107615. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; da Silva Gomes Dias, S.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. SARS-CoV-2 induces inflammasome-dependent pyroptosis and downmodulation of HLA-DR in human monocytes. medRxiv 2020. [Google Scholar] [CrossRef]
- Kucia, M.; Ratajczak, J.; Bujko, K.; Adamiak, M.; Ciechanowicz, A.; Chumak, V.; Brzezniakiewicz-Janus, K.; Ratajczak, M.Z. An evidence that SARS-CoV-2/COVID-19 spike protein (SP) damages hematopoietic stem/progenitor cells in the mechanism of pyroptosis in Nlrp3 inflammasome-dependent manner. Leukemia 2021, 35, 3026–3029. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, A.C.; Mouktaroudi, M.; Krämer, B.; Oestreich, M.; Antonakos, N.; Nuesch-Germano, M.; Gkizeli, K.; Bonaguro, L.; Reusch, N.; Baßler, K.; et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. 2021, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Arora, T.; Wang, X.; Mendelsohn, L.; Nichols, J.; Allen, D.; Shet, A.S.; Combs, C.A.; Quezado, Z.M.N.; Thein, S.L. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018, 2, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.M.; Wanderley, C.W.S.; Veras, F.P.; Gonçalves, A.V.; Lima, M.H.F.; Toller Kawahisa, J.E.; Gomes, G.F.; Nascimento, D.C.; Silva Monteiro, V.V.; Paiva, I.M.; et al. Gasdermin-D activation by SARS-CoV-2 trigger NET and mediate COVID-19 immunopathology. medRxiv 2022. [Google Scholar] [CrossRef]
- Kibbe, M.; Billiar, T.; Tzeng, E. Inducible nitric oxide synthase and vascular injury. Cardiovasc. Res. 1999, 43, 650–657. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Sachdev, V. Cardiovascular abnormalities in sickle cell disease. J. Am. Coll. Cardiol. 2012, 59, 1123–1133. [Google Scholar] [CrossRef]
- Miller, A.C.; Gladwin, M.T. Pulmonary complications of sickle cell disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Miller, M.J.S.; Joshi, M.S.; Sadowska-Krowicka, H.; Clark, D.A.; Lancaster, J.R. Diffusion-limited Reaction of Free Nitric Oxide with Erythrocytes. J. Biol. Chem. 1998, 273, 18709–18713. [Google Scholar] [CrossRef] [Green Version]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Kyle Mack, A.; Kato, G.J. Sickle cell disease and nitric oxide: A paradigm shift? Int. J. Biochem. Cell Biol. 2006, 38, 1237–1243. [Google Scholar] [CrossRef] [Green Version]
- Gladwin, M.T.; Kato, G.J.; Weiner, D.; Onyekwere, O.C.; Dampier, C.; Hsu, L.; Hagar, R.W.; Howard, T.; Nuss, R.; Okam, M.M.; et al. Nitric Oxide for Inhalation in the Acute Treatment of Sickle Cell Pain Crisis: A Randomized Controlled Trial. JAMA 2011, 305, 893–902. [Google Scholar] [CrossRef]
- Green, S.J. COVID-19 accelerates endothelial dysfunction and nitric oxide deficiency. Microbes Infect. 2020, 22, 149–150. [Google Scholar] [CrossRef]
- Akerström, S.; Gunalan, V.; Keng, C.T.; Tan, Y.J.; Mirazimi, A. Dual effect of nitric oxide on SARS-CoV replication: Viral RNA production and palmitoylation of the S protein are affected. Virology 2009, 395, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Akerström, S.; Mousavi-Jazi, M.; Klingström, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Alvarez, R.A.; Berra, L.; Gladwin, M.T. Home Nitric Oxide Therapy for COVID-19. Am. J. Respir. Crit. Care Med. 2020, 202, 16–20. [Google Scholar] [CrossRef]
- Claudia, R.M.; Frans, A.K.; Lisa, L.; Michael, A.; Nancy, S.; Melinee, S.; Ginny, G.; Lynne, N.; Elliott, P.V. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica 2013, 98, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Longobardo, A.; Montanari, C.; Shulman, R.; Benhalim, S.; Singer, M.; Arulkumaran, N. Inhaled nitric oxide produces minimal improvement in oxygenation in COVID-19 related ARDS. Br. J. Anaesth. 2020, 126, E44–E46. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, D.V. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ask, K.; Bonniaud, P.; Maass, K.; Eickelberg, O.; Margetts, P.J.; Warburton, D.; Groffen, J.; Gauldie, J.; Kolb, M. Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1 but not TGF-beta3. Int. J. Biochem. Cell Biol. 2008, 40, 484–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Schroeder, J.A.; Kapoun, A.M.; Damm, D.; Murphy, A.; Chakravarty, S.; Dugar, S.; Higgins, L.; et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am. J. Respir. Crit. Care Med. 2005, 171, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sakata, R.; Ueno, T.; Sata, M.; Ueno, H. Inhibition of transforming growth factor beta prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 2000, 32, 247–255. [Google Scholar] [CrossRef]
- Fukasawa, H.; Yamamoto, T.; Suzuki, H.; Togawa, A.; Ohashi, N.; Fujigaki, Y.; Uchida, C.; Aoki, M.; Hosono, M.; Kitagawa, M.; et al. Treatment with anti-TGF-beta antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-beta signaling. Kidney Int. 2004, 65, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Invest. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [CrossRef]
- Torres Lde, S.; Okumura, J.V.; da Silva, D.G.; Belini Júnior, É.; de Oliveira, R.G.; Mimura, K.K.; Lobo, C.L.; Oliani, S.M.; Bonini Domingos, C.R. Plasma levels of TGF-β1 in homeostasis of the inflammation in sickle cell disease. Cytokine 2016, 80, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Santiago, R.P.; Carvalho, M.O.S.; Figueiredo, C.V.B.; Fiuza, L.M.; Oliveira, R.M.; Yahouédéhou, S.C.M.A.; Nascimento, V.M.L.; Lyra, I.M.; Araujo-Santos, T.; Luz, N.F.; et al. Associations between TGF-β1 Levels and Markers of Hemolysis, Inflammation, and Tissue Remodeling in Pediatric Sickle Cell Patients. Mediat. Inflamm. 2021, 2021, 4651891. [Google Scholar] [CrossRef]
- Witkowski, M.; Tizian, C.; Ferreira-Gomes, M.; Niemeyer, D.; Jones, T.C.; Heinrich, F.; Frischbutter, S.; Angermair, S.; Hohnstein, T.; Mattiola, I.; et al. Untimely TGFβ responses in COVID-19 limit antiviral functions of NK cells. Nature 2021, 600, 295–301. [Google Scholar] [CrossRef]
- Opene, M.; Kurantsin-Mills, J.; Husain, S.; Ibe, B.O. Sickle erythrocytes and platelets augment lung leukotriene synthesis with downregulation of anti-inflammatory proteins: Relevance in the pathology of the acute chest syndrome. Pulm. Circ. 2014, 4, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Field, J.J.; Kassim, A.; Brandow, A.; Embury, S.H.; Matsui, N.; Wilkerson, K.; Bryant, V.; Zhang, L.; Simpson, P.; DeBaun, M.R. Phase 2 trial of montelukast for prevention of pain in sickle cell disease. Blood Adv. 2020, 4, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Žarković, N.; Łuczaj, W.; Jarocka-Karpowicz, I.; Orehovec, B.; Baršić, B.; Tarle, M.; Kmet, M.; Lukšić, I.; Biernacki, M.; Skrzydlewska, E. Diversified Effects of COVID-19 as a Consequence of the Differential Metabolism of Phospholipids and Lipid Peroxidation Evaluated in the Plasma of Survivors and Deceased Patients upon Admission to the Hospital. Int. J. Mol. Sci. 2022, 23, 11810. [Google Scholar] [CrossRef]
- Archambault, A.S.; Zaid, Y.; Rakotoarivelo, V.; Turcotte, C.; Doré, É.; Dubuc, I.; Martin, C.; Flamand, O.; Amar, Y.; Cheikh, A.; et al. High levels of eicosanoids and docosanoids in the lungs of intubated COVID-19 patients. FASEB J. 2021, 35, e21666. [Google Scholar] [CrossRef]
- Camera, M.; Canzano, P.; Brambilla, M.; Rovati, G.E. Montelukast Inhibits Platelet Activation Induced by Plasma From COVID-19 Patients. Front. Pharmacol. 2022, 13, 784214. [Google Scholar] [CrossRef]
- Kerget, B.; Kerget, F.; Aydın, M.; Karaşahin, Ö. Effect of montelukast therapy on clinical course, pulmonary function, and mortality in patients with COVID-19. J. Med. Virol. 2022, 94, 1950–1958. [Google Scholar] [CrossRef]
- Rucker, D.; Dhamoon, A.S. Physiology, Thromboxane A2; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kurantsin-Mills, J.; Ibe, B.O.; Natta, C.L.; Raj, J.U.; Siegel, R.S.; Lessin, L.S. Elevated urinary levels of thromboxane and prostacyclin metabolities in sickle cell disease reflects activated platelets in the circulation. Br. J. Haematol. 1994, 87, 580–585. [Google Scholar] [CrossRef]
- Foulon, I.; Bachir, D.; Galacteros, F.; Maclouf, J. Increased in vivo production of thromboxane in patients with sickle cell disease is accompanied by an impairment of platelet functions to the thromboxane A2 agonist U46619. Arterioscler. Thromb. A J. Vasc. Biol. 1993, 13, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Ibe, B.O.; Morris, J.; Kurantsin-Mills, J.; Raj, J.U. Sickle erythrocytes induce prostacyclin and thromboxane synthesis by isolated perfused rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997, 272, L597–L602. [Google Scholar] [CrossRef]
- Tantry, U.S.; Bliden, K.P.; Cho, A.; Walia, N.; Dahlen, J.R.; Ens, G.; Traianova, M.; Jerjian, C.; Usman, A.; Gurbel, P.A. First Experience Addressing the Prognostic Utility of Novel Urinary Biomarkers in Patients with COVID-19. Open Forum Infect. Dis. 2021, 8, ofab274. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Westcott, J.Y.; Smith, H.R.; Larsen, G.L. Spectrum of prostanoid release after bronchoalveolar allergen challenge in atopic asthmatics and in control groups. An alteration in the ratio of bronchoconstrictive to bronchoprotective mediators. Am. Rev. Respir. Dis. 1989, 139, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.K.; Liu, X.-D.; Chang, H.-Y.; Nagel, R.L.; Fabry, M.E. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. J. Clin. Investig. 2004, 114, 1136–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Garcia, G., Jr.; Wang, Y.; Plummer, J.T.; Morizono, K.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes Are Susceptible to SARS-CoV-2 Infection. Cell Rep. Med. 2020, 1, 100052. [Google Scholar] [CrossRef] [PubMed]
- Ricke-Hoch, M.; Stelling, E.; Lasswitz, L.; Gunesch, A.P.; Kasten, M.; Zapatero-Belinchón, F.J.; Brogden, G.; Gerold, G.; Pietschmann, T.; Montiel, V.; et al. Impaired immune response mediated by prostaglandin E2 promotes severe COVID-19 disease. PLoS ONE 2021, 16, e0255335. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [Green Version]
- Pillinger, M.H.; Capodici, C.; Rosenthal, P.; Kheterpal, N.; Hanft, S.; Philips, M.R.; Weissmann, G. Modes of action of aspirin-like drugs: Salicylates inhibit erk activation and integrin-dependent neutrophil adhesion. Proc. Natl. Acad. Sci. USA 1998, 95, 14540–14545. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, T.; Kawakami, M.; Hidaka, T.; Matsuki, Y.; Takamizawa, M.; Suzuki, K.; Kurita, A.; Nakamura, H. Stimulation with thromboxane A2 (TXA2) receptor agonist enhances ICAM-1, VCAM-1 or ELAM-1 expression by human vascular endothelial cells. Clin. Exp. Immunol. 1998, 112, 464–470. [Google Scholar] [CrossRef]
- Matsui, Y.; Amano, H.; Ito, Y.; Eshima, K.; Suzuki, T.; Ogawa, F.; Iyoda, A.; Satoh, Y.; Kato, S.; Nakamura, M.; et al. Thromboxane A2 receptor signaling facilitates tumor colonization through P-selectin-mediated interaction of tumor cells with platelets and endothelial cells. Cancer Sci. 2012, 103, 700–707. [Google Scholar] [CrossRef]
- Bode, M.; Mackman, N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane A2 as a new player. Vasc. Pharmacol. 2014, 62, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Del Turco, S.; Basta, G.; Lazzerini, G.; Chancharme, L.; Lerond, L.; De Caterina, R. Involvement of the TP receptor in TNF-alpha-induced endothelial tissue factor expression. Vascul. Pharmacol. 2014, 62, 49–56. [Google Scholar] [CrossRef]
- Eligini, S.; Violi, F.; Banfi, C.; Barbieri, S.; Brambilla, M.; Saliola, M.; Tremoli, E.; Colli, S. Indobufen inhibits tissue factor in human monocytes through a thromboxane-mediated mechanism. Cardiovasc. Res. 2006, 69, 218–226. [Google Scholar] [CrossRef]
- Santilli, F.; Davì, G.; Consoli, A.; Cipollone, F.; Mezzetti, A.; Falco, A.; Taraborelli, T.; Devangelio, E.; Ciabattoni, G.; Basili, S.; et al. Thromboxane-Dependent CD40 Ligand Release in Type 2 Diabetes Mellitus. J. Am. Coll. Cardiol. 2006, 47, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-R.; Zhu, Y.; Halushka, P.V.; Lincoln, T.M.; Mendelsohn, M.E. Mechanism of platelet inhibition by nitric oxide: In vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 4888–4893. [Google Scholar] [CrossRef] [Green Version]
- Shiokoshi, T.; Ohsaki, Y.; Kawabe, J.; Fujino, T.; Kikuchi, K. Downregulation of nitric oxide accumulation by cyclooxygenase-2 induction and thromboxane A2 production in interleukin-1β-stimulated rat aortic smooth muscle cells. J. Hypertens. 2002, 20, 455–461. [Google Scholar] [CrossRef]
- Craven, P.A.; Studer, R.K.; DeRubertis, F.R. Thromboxane/Prostaglandin Endoperoxide-Induced Hypertrophy of Rat Vascular Smooth Muscle Cells Is Signaled by Protein Kinase C-Dependent Increases in Transforming Growth Factor-β. Hypertension 1996, 28, 169–176. [Google Scholar] [CrossRef]
- Negrete, H.; Studer, R.K.; Craven, P.A.; DeRubertis, F.R. Role for transforming growth factor beta in thromboxane-induced increases in mesangial cell fibronectin synthesis. Diabetes 1995, 44, 335–339. [Google Scholar] [CrossRef]
- D’Agostino, I.; Tacconelli, S.; Bruno, A.; Contursi, A.; Mucci, L.; Hu, X.; Xie, Y.; Chakraborty, R.; Jain, K.; Sacco, A.; et al. Low-dose Aspirin prevents hypertension and cardiac fibrosis when thromboxane A2 is unrestrained. Pharmacol. Res. 2021, 170, 105744. [Google Scholar] [CrossRef]
- Wood, K.; Gladwin, M.; Straub, A. Sickle cell disease: At the crossroads of pulmonary hypertension and diastolic heart failure. Heart 2019, 106, 562–568. [Google Scholar] [CrossRef]
- Gordeuk, V.R.; Castro, O.L.; Machado, R.F. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016, 127, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A.K.; Hagfjärd, A.; Dahlén, S.E.; Adner, M. Prostaglandin D2 induces contractions through activation of TP receptors in peripheral lung tissue from the guinea pig. Eur. J. Pharmacol. 2011, 669, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Schuster, D.P.; Kozlowski, J.; Brimiouelle, S. Effect of thromboxane receptor blockade on pulmonary capillary hypertension in acute lung injury. In Proceedings of the 2001 Meeting of the American Thoracic Society, San Francisco, CA, USA, 18–23 May 2001. [Google Scholar]
- Carrier, E.J.; Kim, K.; Noll, N.A.; Macias-Perez, I.; Merryman, W.D.; Knollmann, B.C.; West, J.D. Abstract 261: Activation of the Thromboxane/Prostanoid Receptor Contributes to Elevated End-Diastolic Calcium in Cardiomyocytes and Cardiac Fibrosis Following Right Ventricular Pressure Overload. Circ. Res. 2018, 123, A261. [Google Scholar] [CrossRef]
- An, J.; Li, J.Q.; Wang, T.; Li, X.O.; Guo, L.L.; Wan, C.; Liao, Z.L.; Dong, J.J.; Xu, D.; Wen, F.Q. Blocking of thromboxane A(2) receptor attenuates airway mucus hyperproduction induced by cigarette smoke. Eur. J. Pharmacol. 2013, 703, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Horikami, D.; Omori, K.; Nakamura, T.; Yamazaki, A.; Maeda, S.; Murata, T. Thromboxane A2 exacerbates acute lung injury via promoting edema formation. Sci. Rep. 2016, 6, 32109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogletree, M.L.; Chiang, K.C.; Kulshreshta, R.; Agarwal, A.; Agarwal, A.; Gupta, A. Treatment of COVID-19 pneumonia and acute respiratory distress with ramatroban, a thromboxane A2 and prostaglandin D2 receptor antagonist: A 4-Patient Case Series Report. Res. Sq. Platf. LLC 2021. [Google Scholar] [CrossRef]
- Nath, K.A.; Hebbel, R.P. Sickle cell disease: Renal manifestations and mechanisms. Nat. Rev. Nephrol. 2015, 11, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Audard, V.; Bartolucci, P.; Stehlé, T. Sickle cell disease and albuminuria: Recent advances in our understanding of sickle cell nephropathy. Clin. Kidney J. 2017, 10, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Platt, O.S.; Brambilla, D.J.; Rosse, W.F.; Milner, P.F.; Castro, O.; Steinberg, M.H.; Klug, P.P. Mortality In Sickle Cell Disease—Life Expectancy and Risk Factors for Early Death. N. Engl. J. Med. 1994, 330, 1639–1644. [Google Scholar] [CrossRef]
- Hamideh, D.; Alvarez, O. Sickle cell disease related mortality in the United States (1999-2009). Pediatr. Blood Cancer 2013, 60, 1482–1486. [Google Scholar] [CrossRef]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D.; Abate, M.; et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020, 98, 209–218. [Google Scholar] [CrossRef]
- Chan, L.; Coca, S.G. Acute Kidney Injury in the Time of COVID-19. Kidney360 2020, 1, 588–590. [Google Scholar] [CrossRef]
- Gupta, S.; Coca, S.G.; Chan, L.; Melamed, M.L.; Brenner, S.K.; Hayek, S.S.; Sutherland, A.; Puri, S.; Srivastava, A.; Leonberg-Yoo, A.; et al. AKI Treated with Renal Replacement Therapy in Critically Ill Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 161–176. [Google Scholar] [CrossRef]
- Lau, W.L.; Zuckerman, J.E.; Gupta, A.; Kalantar-Zadeh, K. The COVID-Kidney Controversy: Can SARS-CoV-2 Cause Direct Renal Infection? Nephron 2021, 145, 275–279. [Google Scholar] [CrossRef]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Tonneijck, L.; Muskiet, M.H.A.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.L.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- Uriu, K.; Kaizu, K.; Hashimoto, O.; Komine, N.; Etoh, S. Acute and chronic effects of thromboxane A2 inhibition on the renal hemodynamics in streptozotocin-induced diabetic rats. Kidney Int. 1994, 45, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Esmatjes, E.; Conget, I.; Gaya, J.; Fernandez, M.; Ferrer, J.; Rivera-Fillat, F.; Vilardell, E. Effects of Thromboxane Synthesis Inhibitor Triflusal on Renal Hemodynamics in Microalbuminuric Diabetic Patients. Diabetes Care 1990, 13, 1114–1117. [Google Scholar] [CrossRef]
- Bresnahan, B.A.; Dufek, S.; Wu, S.; Lianos, E.A. Changes in glomerular thromboxane A2 receptor expression and ligand binding following immune injury. Kidney Int. 1999, 55, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Loutzenhiser, R.; Epstein, M. Direct evidence that thromboxane mimetic U44069 preferentially constricts the afferent arteriole. J. Am. Soc. Nephrol. 1997, 8, 25–31. [Google Scholar] [CrossRef]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Chadebech, P.; Bodivit, G.; Vieira-Martins, P.; Grunenwald, A.; Boudhabhay, I.; Poillerat, V.; Pakdaman, S.; Kiger, L.; Jouard, A.; et al. Complement activation in sickle cell disease: Dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am. J. Hematol. 2020, 95, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Driss, A.; Wilson, N.O.; Mason, K.; Hyacinth, H.I.; Hibbert, J.M.; Serjeant, G.R.; Stiles, J.K. Elevated IL-1α and CXCL10 serum levels occur in patients with homozygous sickle cell disease and a history of acute splenic sequestration. Dis. Markers 2012, 32, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Bustos, M.; Coffman, T.M.; Saadi, S.; Platt, J.L. Modulation of eicosanoid metabolism in endothelial cells in a xenograft model. Role of cyclooxygenase-2. J. Clin. Investig. 1997, 100, 1150–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Holter, J.C.; Pischke, S.E.; De Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef]
- England, H.; Summersgill, H.R.; Edye, M.E.; Rothwell, N.J.; Brough, D. Release of Interleukin-1α or Interleukin-1β Depends on Mechanism of Cell Death. J. Biol. Chem. 2014, 289, 15942–15950. [Google Scholar] [CrossRef] [Green Version]
- Böhm, E.; Sturm, G.J.; Weiglhofer, I.; Sandig, H.; Shichijo, M.; McNamee, A.; Pease, J.E.; Kollroser, M.; Peskar, B.A.; Heinemann, A. 11-Dehydro-thromboxane B2, a Stable Thromboxane Metabolite, Is a Full Agonist of Chemoattractant Receptor-homologous Molecule Expressed on TH2 Cells (CRTH2) in Human Eosinophils and Basophils. J. Biol. Chem. 2004, 279, 7663–7670. [Google Scholar] [CrossRef] [Green Version]
- Westlund, P.; Kumlin, M.; Nordenström, A.; Granström, E. Circulating and urinary thromboxane B2 metabolites in the rabbit: 11-dehydro-thromboxane B2 as parameter of thromboxane production. Prostaglandins 1986, 31, 413–443. [Google Scholar] [CrossRef]
- Spellberg, B.; Edwards, J.E. Type 1/Type 2 Immunity in Infectious Diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef] [Green Version]
- Roncati, L.; Nasillo, V.; Lusenti, B.; Riva, G. Signals of Th2 immune response from COVID-19 patients requiring intensive care. Ann. Hematol. 2020, 99, 1419–1420. [Google Scholar] [CrossRef]
- Domingo, C.; Palomares, O.; Sandham, D.A.; Erpenbeck, V.J.; Altman, P. The prostaglandin D2 receptor 2 pathway in asthma: A key player in airway inflammation. Respir. Res. 2018, 19, 189. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Donlan, A.N.; Sutherland, T.E.; Marie, C.; Preissner, S.; Bradley, B.T.; Carpenter, R.M.; Sturek, J.M.; Ma, J.Z.; Moreau, G.B.; Donowitz, J.R.; et al. IL-13 is a driver of COVID-19 severity. JCI Insight 2021, 6, e150107. [Google Scholar] [CrossRef]
- Gómez-Escobar, L.G.; Hoffman, K.L.; Choi, J.J.; Borczuk, A.; Salvatore, S.; Alvarez-Mulett, S.L.; Galvan, M.D.; Zhao, Z.; Racine-Brzostek, S.E.; Yang, H.S.; et al. Cytokine signatures of end organ injury in COVID-19. Sci. Rep. 2021, 11, 12606. [Google Scholar] [CrossRef]
- Arima, M.; Fukuda, T. Prostaglandin D2and TH2 Inflammation in the Pathogenesis of Bronchial Asthma. Korean J. Intern. Med. 2011, 26, 8. [Google Scholar] [CrossRef]
- Xue, L.; Gyles, S.L.; Wettey, F.R.; Gazi, L.; Townsend, E.; Hunter, M.G.; Pettipher, R. Prostaglandin D2 Causes Preferential Induction of Proinflammatory Th2 Cytokine Production through an Action on Chemoattractant Receptor-Like Molecule Expressed on Th2 Cells. J. Immunol. 2005, 175, 6531–6536. [Google Scholar] [CrossRef] [Green Version]
- Trabanelli, S.; Chevalier, M.F.; Martinez-Usatorre, A.; Gomez-Cadena, A.; Salome, B.; Lecciso, M.; Salvestrini, V.; Verdeil, G.; Racle, J.; Papayannidis, C.; et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun. 2017, 8, 593. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, H.; Luo, M.; Liu, J.; Wu, L.; Lin, X.; Li, R.; Wang, Z.; Zhong, H.; Zheng, W.; et al. Lymphopenia predicted illness severity and recovery in patients with COVID-19: A single-center, retrospective study. PLoS ONE 2020, 15, e0241659. [Google Scholar] [CrossRef]
- Abadpour, S.; Tyrberg, B.; Schive, S.W.; Huldt, C.W.; Gennemark, P.; Ryberg, E.; Rydén-Bergsten, T.; Smith, D.M.; Korsgren, O.; Skrtic, S.; et al. Inhibition of the prostaglandin D2–GPR44/DP2 axis improves human islet survival and function. Diabetologia 2020, 63, 1355–1367. [Google Scholar] [CrossRef]
- Zuo, S.; Kong, D.; Wang, C.; Liu, J.; Wang, Y.; Wan, Q.; Yan, S.; Zhang, J.; Tang, J.; Zhang, Q.; et al. CRTH2 promotes endoplasmic reticulum stress-induced cardiomyocyte apoptosis through m-calpain. EMBO Mol. Med. 2018, 10, e8237. [Google Scholar] [CrossRef]
- Yue, L.; Durand, M.; Lebeau Jacob, M.C.; Hogan, P.; McManus, S.; Roux, S.; de Brum-Fernandes, A.J. Prostaglandin D2 induces apoptosis of human osteoclasts by activating the CRTH2 receptor and the intrinsic apoptosis pathway. Bone 2012, 51, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 2006, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Debaun, M.R.; Jordan, L.C.; King, A.A.; Schatz, J.; Vichinsky, E.; Fox, C.K.; McKinstry, R.C.; Telfer, P.; Kraut, M.A.; Daraz, L.; et al. American Society of Hematology 2020 guidelines for sickle cell disease: Prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020, 4, 1554–1588. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.P.; Streiff, M.B.; Lanzkron, S. Sickle cell disease and venous thromboembolism: What the anticoagulation expert needs to know. J. Thromb. Thrombolysis 2013, 35, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biemond, B.J.; Tombak, A.; Kilinc, Y.; Al-Khabori, M.; Abboud, M.; Nafea, M.; Inati, A.; Wali, Y.; Kristensen, J.; Kowalski, J.; et al. Sevuparin for the treatment of acute pain crisis in patients with sickle cell disease: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Haematol. 2021, 8, e334–e343. [Google Scholar] [CrossRef] [PubMed]
- Qari, M.H.; Aljaouni, S.K.; Alardawi, M.S.; Fatani, H.; Alsayes, F.M.; Zografos, P.; Alsaigh, M.; Alalfi, A.; Alamin, M.; Gadi, A.; et al. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb. Haemost. 2007, 98, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.; Ohene-Frempong, K.; Halus, J.; Way, C.; Schwartz, E. Trial of low doses of aspirin as prophylaxis in sickle cell disease. J. Pediatr. 1983, 102, 781–784. [Google Scholar] [CrossRef]
- Heeney, M.M.; Hoppe, C.C.; Abboud, M.R.; Inusa, B.; Kanter, J.; Ogutu, B.; Brown, P.B.; Heath, L.E.; Jakubowski, J.A.; Zhou, C.; et al. A Multinational Trial of Prasugrel for Sickle Cell Vaso-Occlusive Events. N. Engl. J. Med. 2016, 374, 625–635. [Google Scholar] [CrossRef]
- Kanter, J.; Abboud, M.R.; Kaya, B.; Nduba, V.; Amilon, C.; Gottfridsson, C.; Rensfeldt, M.; Leonsson-Zachrisson, M.; HESTIA2 Study Investigators. Ticagrelor does not impact patient-reported pain in young adults with sickle cell disease: A multicentre, randomised phase IIb study. Br. J. Haematol. 2019, 184, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Warlo, E.M.K.; Arnesen, H.; Seljeflot, I. A brief review on resistance to P2Y12 receptor antagonism in coronary artery disease. Thromb. J. 2019, 17, 11. [Google Scholar] [CrossRef]
- Desai, P.C.; Brittain, J.E.; Jones, S.K.; McDonald, A.; Wilson, D.R.; Dominik, R.; Key, N.S.; Parise, L.V.; Ataga, K.I. A pilot study of eptifibatide for treatment of acute pain episodes in sickle cell disease. Thromb. Res. 2013, 132, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Rees, D.C.; Kilinc, Y.; Unal, S.; Dampier, C.; Pace, B.S.; Kaya, B.; Trompeter, S.; Odame, I.; Mahlangu, J.; Unal, S.; et al. A randomized, placebo-controlled, double-blind trial of canakinumab in children and young adults with sickle cell anemia. Blood 2022, 139, 2642–2652. [Google Scholar] [CrossRef]
- NIH. Antithrombotic Therapy in Patients with COVID-19. Available online: https://www.covid19treatmentguidelines.nih.gov/adjunctive-therapy/antithrombotic-therapy/ (accessed on 5 December 2022).
- Phend, C. Trials Halt Full-Dose Clot Prophylaxis for Severe COVID-19. Available online: https://www.medpagetoday.com/infectiousdisease/covid19/90351 (accessed on 1 January 2022).
- Daviet, F.; Guervilly, C.; Baldesi, O.; Bernard-Guervilly, F.; Pilarczyk, E.; Genin, A.; Lefebvre, L.; Forel, J.M.; Papazian, L.; Camoin-Jau, L. Heparin-Induced Thrombocytopenia in Severe COVID-19. Circulation 2020, 142, 1875–1877. [Google Scholar] [CrossRef]
- Hughes, S. COVID-19 Anticoagulation Trials ‘Paused’ for Futility, Safety. Available online: https://www.medscape.com/viewarticle/943085 (accessed on 1 January 2022).
- Connors, J.M.; Brooks, M.M.; Sciurba, F.C.; Krishnan, J.A.; Bledsoe, J.R.; Kindzelski, A.; Baucom, A.L.; Kirwan, B.-A.; Eng, H.; Martin, D.; et al. Effect of Antithrombotic Therapy on Clinical Outcomes in Outpatients with Clinically Stable Symptomatic COVID-19: The ACTIV-4B Randomized Clinical Trial. JAMA 2021, 326, 1703–1712. [Google Scholar] [CrossRef]
- Osborne, T.F.; Veigulis, Z.P.; Arreola, D.M.; Mahajan, S.M.; Röösli, E.; Curtin, C.M. Association of mortality and aspirin prescription for COVID-19 patients at the Veterans Health Administration. PLoS ONE 2021, 16, e0246825. [Google Scholar] [CrossRef]
- RECOVERY. Aspirin to be Investigated as a Possible Treatment for COVID-19 in the RECOVERY Trial. Available online: https://www.recoverytrial.net/news/aspirin-to-be-investigated-as-a-possible-treatment-for-covid-19-in-the-recovery-trial (accessed on 8 December 2022).
- Horby, P.W.; Pessoa-Amorim, G.; Staplin, N.; Emberson, J.R.; Campbell, M.; Spata, E.; Peto, L.; Brunskill, N.J.; Tiberi, S.; Chew, V.; et al. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. medRxiv 2021. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Boeglin, W.E.; Boutaud, O.; Malkowski, M.G.; Schneider, C. Residual cyclooxygenase activity of aspirin-acetylated COX-2 forms 15 R-prostaglandins that inhibit platelet aggregation. FASEB J. 2019, 33, 1033–1041. [Google Scholar] [CrossRef]
- Kim, J.W.; Zou, Y.; Yoon, S.; Lee, J.H.; Kim, Y.K.; Yu, B.P.; Chung, H.Y. Vascular aging: Molecular modulation of the prostanoid cascade by calorie restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, B876–B885. [Google Scholar] [CrossRef] [Green Version]
- Petrucci, G.; Zaccardi, F.; Giaretta, A.; Cavalca, V.; Capristo, E.; Cardillo, C.; Pitocco, D.; Porro, B.; Schinzari, F.; Toffolo, G.; et al. Obesity is associated with impaired responsiveness to once-daily low-dose aspirin and in vivo platelet activation. J. Thromb. Haemost. 2019, 17, 885–895. [Google Scholar] [CrossRef]
- Eikelboom, J.W.; Hankey, G.J.; Thom, J.; Bhatt, D.L.; Steg, P.G.; Montalescot, G.; Johnston, S.C.; Steinhubl, S.R.; Mak, K.-H.; Easton, J.D.; et al. Incomplete Inhibition of Thromboxane Biosynthesis by Acetylsalicylic Acid. Circulation 2008, 118, 1705–1712. [Google Scholar] [CrossRef] [Green Version]
- Petito, E.; Falcinelli, E.; Paliani, U.; Cesari, E.; Vaudo, G.; Sebastiano, M.; Cerotto, V.; Guglielmini, G.; Gori, F.; Malvestiti, M.; et al. Association of Neutrophil Activation, more than Platelet Activation, with Thrombotic Complications in Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S. Aspirin use to be banned in under 16 year olds. BMJ 2002, 325, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capuano, A.; Scavone, C.; Racagni, G.; Scaglione, F. NSAIDs in patients with viral infections, including COVID-19: Victims or perpetrators? Pharmacol. Res. 2020, 157, 104849. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Can COX-2 inhibitor-induced increase in cardiovascular disease risk be modified by essential fatty acids? J. Assoc. Physicians India 2005, 53, 623–627. [Google Scholar]
- Antman, E.M.; DeMets, D.; Loscalzo, J. Cyclooxygenase Inhibition and Cardiovascular Risk. Circulation 2005, 112, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Patscheke, H.; Hornberger, W.; Zehender, H. Pathophysiological role of thromboxane A2 and pharmacological approaches to its inhibition. Z Kardiol. 1990, 79 Suppl 3, 151–154. [Google Scholar]
- Gupta, A.; Kalantar-Zadeh, K.; Srinivasa, R.T. Ramatroban as a Novel Immunotherapy for COVID-19. Mol. Genet. Med. 2020, 14, 1–4. [Google Scholar] [CrossRef]
- Pang, J.; Qi, X.; Luo, Y.; Li, X.; Shu, T.; Li, B.; Song, M.; Liu, Y.; Wei, D.; Chen, J.; et al. Multi-omics study of silicosis reveals the potential therapeutic targets PGD(2) and TXA(2). Theranostics 2021, 11, 2381–2394. [Google Scholar] [CrossRef]
- Ishizuka, T.; Matsui, T.; Okamoto, Y.; Ohta, A.; Shichijo, M. Ramatroban (BAY u 3405): A novel dual antagonist of TXA2 receptor and CRTh2, a newly identified prostaglandin D2 receptor. Cardiovasc. Drug Rev. 2004, 22, 71–90. [Google Scholar] [CrossRef]
- Kariyazono, H.; Nakamura, K.; Arima, J.; Ayukawa, O.; Onimaru, S.; Masuda, H.; Iguro, Y.; Majima, H.J.; Sakata, R.; Yamada, K. Evaluation of anti-platelet aggregatory effects of aspirin, cilostazol and ramatroban on platelet-rich plasma and whole blood. Blood Coagul. Fibrinolysis 2004, 15, 157–167. [Google Scholar] [CrossRef]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nüsing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schrör, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011, 9, 790–798. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.; Bornikova, L.; Gupta, S.; et al. COVID and Coagulation: Bleeding and Thrombotic Manifestations of SARS-CoV-2 Infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Christen, J.R.; Bertolino, J.; Jean, E.; Camoin, L.; Ebbo, M.; Harlé, J.R.; Schleinitz, N.; Sarlon, G.; Bernit, E. Use of Direct Oral Anticoagulants in Patients with Sickle Cell Disease and Venous Thromboembolism: A Prospective Cohort Study of 12 Patients. Hemoglobin 2019, 43, 296–299. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Subjects | Age/Reference | Plasma D-Dimer Levels | p-Value Compared to Controls | |
|---|---|---|---|---|
| Control | Disease State | |||
| Sickle Cell Disease | Adult [76] | HD (n = 35) 79 ± 25 ngh | Steady State SCD n = 25 (Samples = 28) 566 ± 739 ng/mL | p < 0.001 |
| SCD Painful Crisis n = 21 (Samples = 40) 1038 ± 1010 ng/mL | p < 0.001 | |||
| 12–37 years [75] | SCD with pain crisis and normal chest X-ray (n episodes = 32) 584.2 µg/L (250–3119 µg/L) | SCD with pain crisis and abnormal chest X-ray (n episodes = 13) 2117.0 µg/L (250–9143 µg/L) | N/A | |
| Unventilated: 62.5 ± 8.4 Ventilated: 53.8 ± 9.3 [77] | Hospitalized COVID-19 patients did not require artificial ventilation (n = 18) 650 ± 175 ng/mL | Hospitalized COVID-19 patients requiring artificial ventilation (n = 11) 1250 ± 210 ng/mL | p < 0.05 | |
| COVID-19 | 65.57 ± 13 years [78] | COVID-19 patients without pulmonary embolism (n = 118) 1310 ng/mL (800–2335) | COVID-19 patients with pulmonary embolism (n = 44) 5364 ng/mL (2928–12,275) | p = 0.001 |
| Subjects | Source and Analyte | Thromboxane Levels | p-Value Compared to Controls | |
|---|---|---|---|---|
| Control | Disease State | |||
| Sickle Cell Disease | Plasma 2,3 dinor-TxB2 [140] (µg/L) (Mean ± SEM) | HD (n = 12) 2.75 ± 0.83 | Steady State SCD (n = 15) 21.53 ± 5.10 | p < 0.001 |
| Plasma TxB2 [140] (µg/L) (Mean ± SEM) | HD (n = 12) <0.005 | Steady State SCD (n = 15) 0.543 ± 0.101 | p < 0.05 | |
| Urinary TxB2 [140] (pg/mg creatinine) (Mean ± SEM) | HD (n = 12) 0.41 ± 0.30 | Steady State SCD (n = 15) 0.91 ± 0.13 | p < 0.05 | |
| Urinary 2,3 dinor-TxB2 [140] (pg/mg creatinine) (mean ± SEM) | HD (n = 12) 1.70 ± 0.032 | Steady State SCD (n = 15) 2.81 ± 0.13 | p < 0.01 | |
| Urinary 11-dehydro-TxB2 [141] (pg/mg creatinine) (Mean ± SEM) | HD (n = 33) 299 ± 20 | Steady State SCD (n = 49) 1227 ± 191 | p = 0.0002 | |
| Vaso-Occlusive SCD (n = 15) 1836 ± 536 | p = 0.0005 | |||
| COVID-19 | BALF TxB2 [52] (nmol/L) (Means) | HD (n = 25) <0.250 | Severe COVID-19 (n = 33) 12.0 | p < 0.0001 |
| Plasma TxB2 [23] (ng/mL) (Median) | HD (n = 11) 4.0 | Severe COVID-19 (n = 35) 7.5 | p < 0.05 | |
| Urinary 11-dehydro-TxB2 [143] (pg/mg creatinine) (Median (95% CI)) | Without Events (n = 47) 4890 (5049–8290) | With Events (n = 18) 7603 (7541–19,791) | p = 0.002 | |
| <10 d of hospitalization (n = 35) 4801 (3817–9196) | ≥10 d of hospitalization (n = 30) 8614 (7990–14,316) | p = 0.02 | ||
| No death (n = 48) 5360 (5907–10,038) | Death (n = 6) 15,069 (1915–42,007) | p = 0.004 | ||
| No Mechanical Ventilation (n = 56) 5137 (4498–7512) | Mechanical Ventilation (n = 9) 20,121 (5364–41,015) | p < 0.001 | ||
| Atopic Asthmatics | BALF TxB2 [144] (nmol/L) (Mean ± SEM) | Before Allergen Challenge (n = 8) 0.130 ± 0.021 | After Allergen Challenge (n = 8) 0.430 ± 0.108 | p < 0.05 |
| Study Design | Study Population | Intervention | Primary Outcome Measure and Result |
|---|---|---|---|
| Phase IIb Multicenter, double-blind, double-dummy, randomized, placebo-controlled, parallel-group [213] | ● Have SCD [homozygous sickle cell (HbSS) or sickle beta-zero thalassemia (HbSβ0)] ● Ages 18–30 years (mean 22.2 years old) ● Have ≥4 days of pain during the 4-week single-blind placebo baseline period prior to randomization ● If on hydroxycarbamide, a stable dose for 3 months prior to enrollment required ● If on erythropoietin, drug must have been prescribed 6 months before and at a stable dose for ≥3 months prior to randomization (n = 194) | ● Ticagrelor 10 mg plus matching placebo for ticagrelor 45 mg ● Ticagrelor 45 mg plus matching placebo for ticagrelor 10 mg ● Matching placebo for ticagrelor 10 and 45 mg Duration: 12 weeks | Proportions of days with diary-reported SCD pain No significant difference between placebo and ticagrelor treatment groups |
| Phase III Multinational, double-blind, randomized, placebo-controlled, parallel-group [212] | ● Have SCD [homozygous sickle cell (HbSS) or sickle beta-zero thalassemia (HbSβ0)] ● Are participants with SCD who have had ≥2 episodes of vaso-occlusive crisis (VOC) in the past year ● Have a body weight ≥ 19 kilogram (kg) and are ≥ 2 and <18 years of age, inclusive at the time of screening ● If participants are ≥2 and ≤16 years of age, they must have had a transcranial Doppler within the last year (n = 341) | ● Prasugrel 0.08–0.12 mg/kg po once daily ● Placebo Duration: 9–24 months | Number of Vaso-Occlusive Crisis (VOC) Events Per Participant Per Year (Rate of VOC) Terminated due to lack of efficacy |
| Phase III Double-blind crossover study [211] | ● Have sickle hemoglobinopathy observed regularly ● Ages 2–17 years old (mean 7.7 years old) ● The hematologic diagnosis was confirmed by cellulose acetate electrophoresis at pH 8.6 and citrate agar electrophoresis at pH 6.4 ● At least 50% compliant (n = 49) | ● Low dose aspirin ● Placebo | Frequency and severity of VOC No significant difference between placebo and aspirin treatment groups |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, K.C.; Gupta, A.; Sundd, P.; Krishnamurti, L. Thrombo-Inflammation in COVID-19 and Sickle Cell Disease: Two Faces of the Same Coin. Biomedicines 2023, 11, 338. https://doi.org/10.3390/biomedicines11020338
Chiang KC, Gupta A, Sundd P, Krishnamurti L. Thrombo-Inflammation in COVID-19 and Sickle Cell Disease: Two Faces of the Same Coin. Biomedicines. 2023; 11(2):338. https://doi.org/10.3390/biomedicines11020338
Chicago/Turabian StyleChiang, Kate Chander, Ajay Gupta, Prithu Sundd, and Lakshmanan Krishnamurti. 2023. "Thrombo-Inflammation in COVID-19 and Sickle Cell Disease: Two Faces of the Same Coin" Biomedicines 11, no. 2: 338. https://doi.org/10.3390/biomedicines11020338
APA StyleChiang, K. C., Gupta, A., Sundd, P., & Krishnamurti, L. (2023). Thrombo-Inflammation in COVID-19 and Sickle Cell Disease: Two Faces of the Same Coin. Biomedicines, 11(2), 338. https://doi.org/10.3390/biomedicines11020338