
Development of the Rabbit NASH Model Resembling Human NASH and Atherosclerosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Histological Analysis
2.3. Biochemical Analysis
2.4. Quantification of mRNA Measurement
2.5. Western Blot Analysis
2.6. Statistical Analysis
3. Results
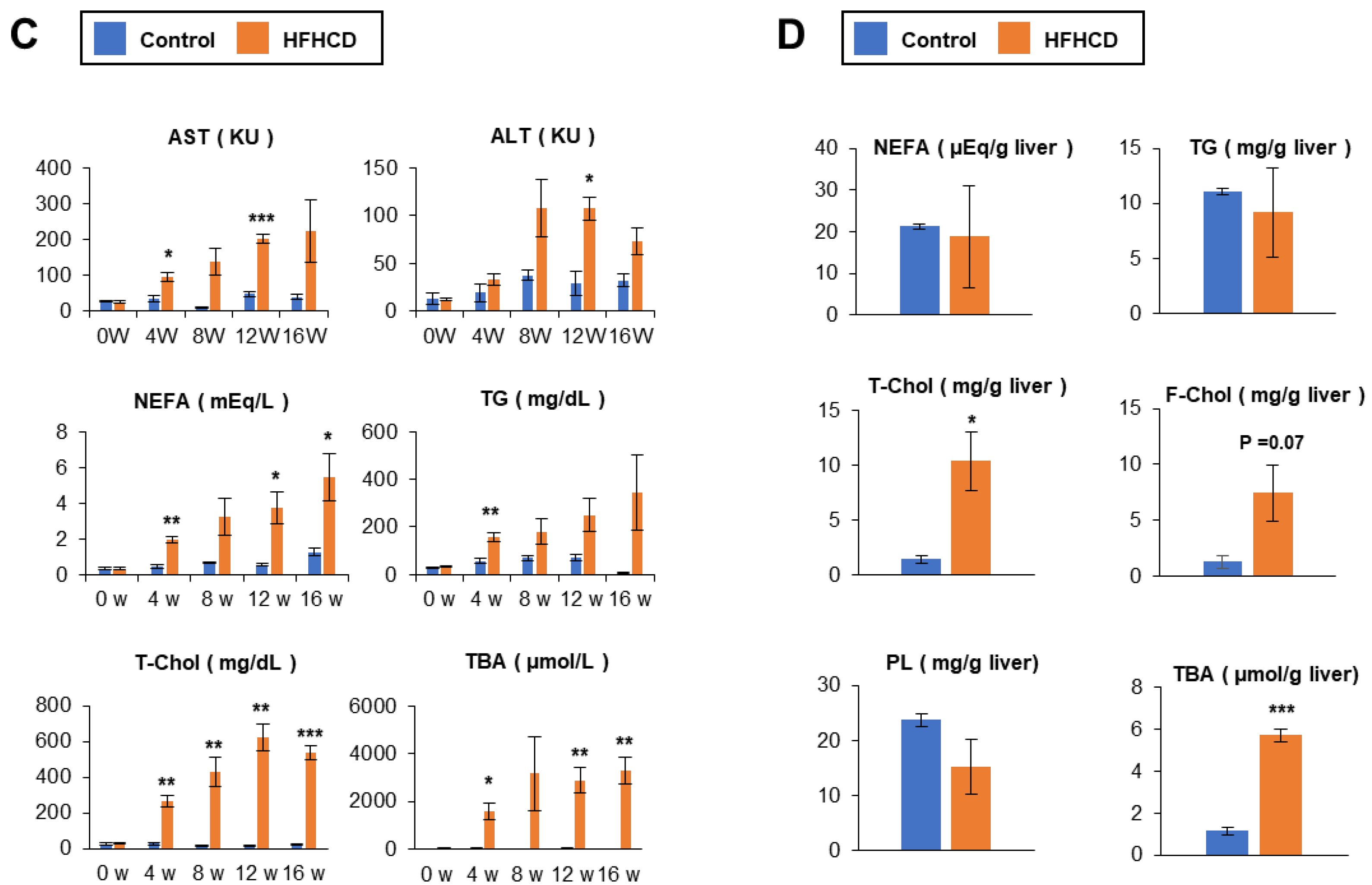
3.1. HFHCD Induces NASH with Advanced Liver Fibrosis and Severe Atherosclerosis in Rabbits for 16 Weeks
3.2. Time–Course Study of HFHCD-fed Rabbits
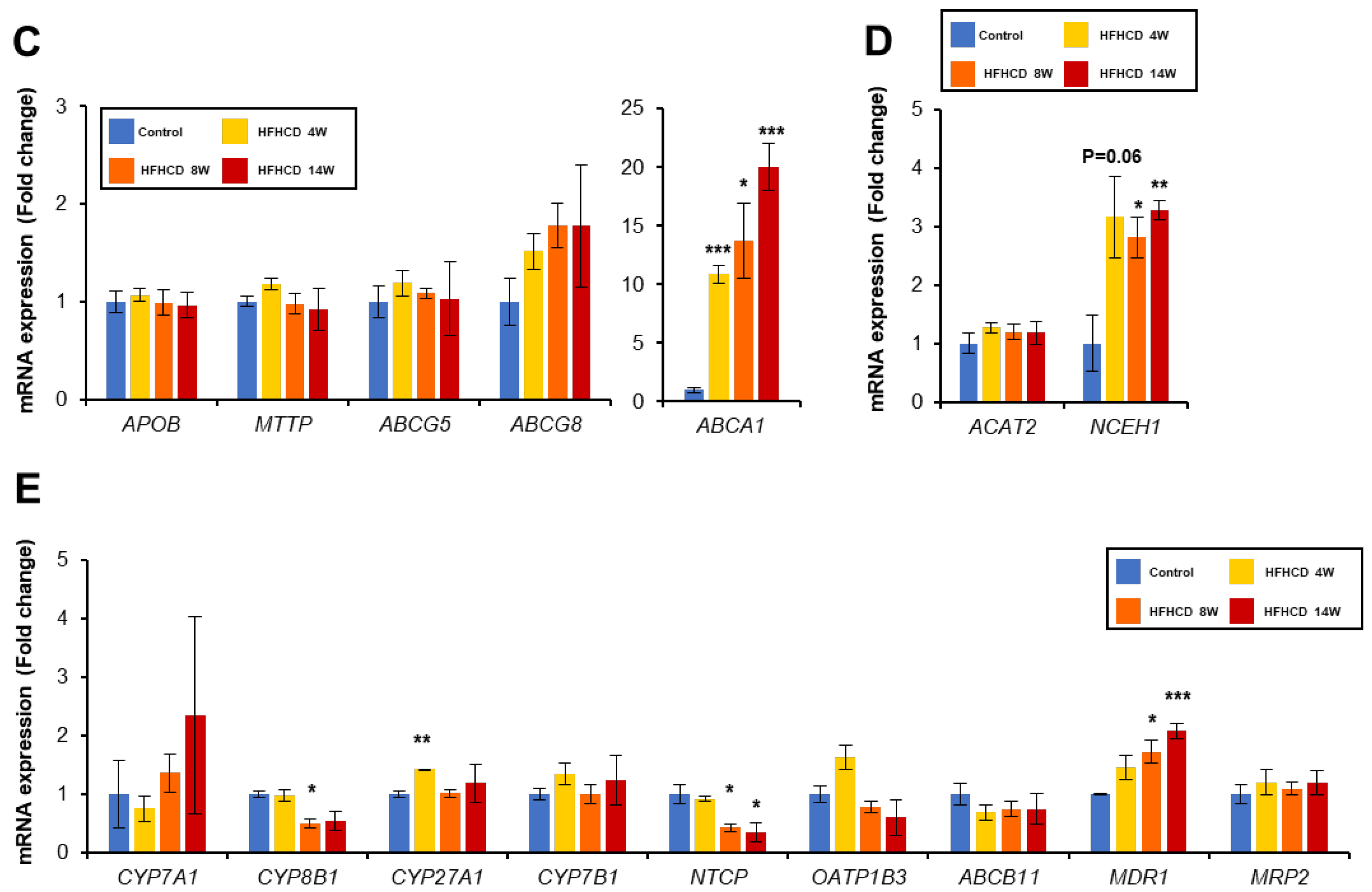
3.3. HFHCD Alters Gene Expression Associated with Cholesterol Metabolism
3.4. HFHCD Disrupts BA Homeostasis in the Liver
3.5. HFHCD Does Not Affect the Expression of Genes Related to Fatty Acid Metabolism
3.6. HFHCD Up-Regulates Gene Expression Related to Inflammation and Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-nonenal |
| ACACA | acetyl-CoA carboxylase |
| ACATA2 | acetyl-CoA acetyltransferase 2 |
| ALT | alanine aminotransferase |
| apoB | apolipoprotein B |
| ASCL1 | long-chain-fatty acid CoA ligase 1 |
| AST | aspartate aminotransferase |
| BA | bile acid |
| BIM | Bcl-2-like protein 11 |
| BSEP | bile salt export pump |
| CA | cholic acid |
| CCL2 | C-C motif chemokine ligand 2 |
| CDCA | chenodeoxycholic acid |
| CETP | cholesterol ester transferase |
| COL1A1 | collagen type I alpha 1 chain |
| COL3A1 | collagen type III alpha 1 chain |
| CVD | cardiovascular disease |
| CYBB | cytochrome b-245 beta chain |
| CYP7A1 | cytochrome P450 7A1 |
| DGAT2 | diacylglycerol acyltransferase 2 |
| ER | endoplasmic reticulum |
| FABP1 | fatty acid-binding protein 1 |
| F-Chol | free cholesterol |
| FDFT1 | farnesyl-diphosphate farnesyltransferase 1 |
| FXR | farnesoid X receptor |
| HDL | high-density lipoprotein |
| HFHCD | high-fat high-cholesterol diet |
| HMGCR | HMG-CoA reductase |
| HMOX1 | heme oxygenase 1 |
| HTGL | hepatic triglyceride lipase |
| LDL | low-density lipoprotein |
| LDLR | low-density lipoprotein receptor |
| MCA | muricholic acid |
| MDR1 | multidrug resistance 1 |
| MI | myocardial infarction |
| MRP2 | multidrug resistance-associated protein 2 |
| MTTP | microsomal triglyceride transfer protein |
| MYD88 | myeloid differentiation primary response 88 |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NCEH1 | neutral cholesterol ester hydrolase 1 |
| NCF1 | neutrophil cytosolic factor 1 |
| NEFA | non-esterified fatty acid |
| NLRP3 | NLR family pyrin domain containing 3 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NTCP | solute carrier family 10 member 1 |
| PERK | PRKR-like endoplasmic reticulum kinase |
| PL | phospholipid |
| PUMA | p53 upregulated modulator of apoptosis |
| RCT | reverse cholesterol transport |
| SOD1 | superoxide dismutase 1 |
| SQLE | squalene monooxygenase |
| SREBP-2 | sterol regulatory element-binding protein-2 |
| TBA | total bile acid |
| T-Chol | total cholesterol |
| TG | triacylglycerol |
| TGFβ | transforming growth factor β |
| TLR | toll-like receptor |
| TNFα | tumor necrosis factor α |
| VLDL | very low-density lipoprotein |
References
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kimura, T.; Fujimori, N.; Nagaya, T.; Komatsu, M.; Tanaka, E. Current status, problems, and perspectives of non-alcoholic fatty liver disease research. World J. Gastroenterol. 2019, 25, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, F.P.; Hu, X.; Kimura, T.; Tanaka, N. Impact of Dietary Fat on the Progression of Liver Fibrosis: Lessons from Animal and Cell Studies. Int. J. Mol. Sci. 2021, 22, 303. [Google Scholar] [CrossRef]
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019, 76, 99–128. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Hirsova, P.; Gores, G.J. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Diao, P.; Wang, X.J.; Jia, F.P.; Kimura, T.; Hu, X.; Shirotori, S.; Nakamura, I.; Sato, Y.; Nakayama, J.; Moriya, K.; et al. A saturated fatty acid-rich diet enhances hepatic lipogenesis and tumorigenesis in HCV core gene transgenic mice. J. Nutr. Biochem. 2020, 85, 108460. [Google Scholar] [CrossRef]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef]
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, X.H.; Ou, X.; Ouyang, X.P.; Tang, C.K. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and atherosclerosis. Prog. Lipid Res. 2021, 83, 101109. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.J.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; She, Z.G.; Li, H.L. Nonalcoholic Fatty Liver Disease Pandemic Fuels the Upsurge in Cardiovascular Diseases. Circ. Res. 2020, 126, 679–704. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Stols-Goncalves, D.; Hovingh, G.K.; Nieuwdorp, M.; Holleboom, A.G. NAFLD and Atherosclerosis: Two Sides of the Same Dysmetabolic Coin? Trends Endocrinol. Metab. 2019, 30, 891–902. [Google Scholar] [CrossRef]
- Wojcik-Cichy, K.; Koslinska-Berkan, E.; Piekarska, A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin. Exp. Hepatol. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Yoshitaka, H.; Hamaguchi, M.; Kojima, T.; Fukuda, T.; Ohbora, A.; Fukui, M. Nonoverweight nonalcoholic fatty liver disease and incident cardiovascular disease A post hoc analysis of a cohort study. Medicine 2017, 96, e6712. [Google Scholar] [CrossRef]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: An updated systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef]
- Tanaka, N.; Matsubara, T.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 2012, 56, 118–129. [Google Scholar] [CrossRef]
- Tanaka, N.; Takahashi, S.; Fang, Z.Z.; Matsubara, T.; Krausz, K.W.; Qu, A.J.; Gonzalez, F.J. Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 1596–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Takahashi, S.; Zhang, Y.; Krausz, K.W.; Smith, P.B.; Patterson, A.D.; Gonzalez, F.J. Role of fibroblast growth factor 21 in the early stage of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Takahashi, S.; Hu, X.; Lu, Y.; Fujimori, N.; Golla, S.; Fang, Z.Z.; Aoyama, T.; Krausz, K.W.; Gonzalez, F.J. Growth arrest and DNA damage-inducible 45 alpha protects against nonalcoholic steatohepatitis induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 3170–3182. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Straniero, S.; Laskar, A.; Savva, C.; Hardfeldt, J.; Angelin, B.; Rudling, M. Of mice and men: Murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J. Lipid Res. 2020, 61, 480–491. [Google Scholar] [CrossRef] [Green Version]
- Li, J.N.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta-Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef]
- Shiomi, M.; Koike, T.; Ishida, T. Genetically Modified Animal Models for Lipoprotein Research. In Lipoproteins—Role in Health and Diseases; Frank, S., Kostner, G., Eds.; IntechOpen: London, UK, 2012. [Google Scholar]
- Shiomi, M. The History of the WHHL Rabbit, an Animal Model of Familial Hypercholesterolemia (II)—Contribution to the Development and Validation of the Therapeutics for Hypercholesterolemia and Atherosclerosis. J. Atheroscler. Thromb. 2020, 27, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Thakare, R.; Alamoudi, J.A.; Gautam, N.; Rodrigues, A.D.; Alnouti, Y. Species differences in bile acids I. Plasma and urine bile acid composition. J. Appl. Toxicol. 2018, 38, 1323–1335. [Google Scholar] [CrossRef]
- Brophy, M.L.; Dong, Y.Z.; Wu, H.; Rahman, H.N.A.; Song, K.; Chen, H. Eating the Dead to Keep Atherosclerosis at Bay. Front. Cardiovasc. Med. 2017, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Veseli, B.E.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Fatima, M.; Hou, S.Y.; Bai, L.; Zhao, S.H.; Liu, E.Q. Research methods for animal models of atherosclerosis. Mol. Med. Rep. 2021, 24. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.L.; Kitajima, S.; Watanabe, T.; Xu, J.; Zhang, J.F.; Liu, E.Q.; Chen, Y.E. Rabbit models for the study of human atherosclerosis: From pathophysiological mechanisms to translational medicine. Pharmacol. Ther. 2015, 146, 104–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Koike, T.; Nakagawa, T.; Kuniyoshi, N.; Ying, Y.; Itabe, H.; Yamashita, A.; Asada, Y.; Shiomi, M. Morphological features of coronary plaques in WHHLMI rabbits (Oryctolagus cuniculus), an animal model for familial hypercholesterolemia. Exp. Anim. 2017, 66, 145–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Tanaka, N.; Guo, R.; Lu, Y.; Nakajima, T.; Gonzalez, F.J.; Aoyama, T. PPAR alpha protects against trans-fatty-acid-containing diet-induced steatohepatitis. J. Nutr. Biochem. 2017, 39, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Tanaka, N.; Hu, X.; Kimura, T.; Lu, Y.; Jia, F.P.; Sato, Y.; Nakayama, J.; Moriya, K.; Koike, K.; et al. A high-cholesterol diet promotes steatohepatitis and liver tumorigenesis in HCV core gene transgenic mice. Arch. Toxicol. 2019, 93, 1713–1725. [Google Scholar] [CrossRef]
- Hu, X.; Wang, X.J.; Jia, F.P.; Tanaka, N.; Kimura, T.; Nakajima, T.; Sato, Y.; Moriya, K.; Koike, K.; Gonzalez, F.J.; et al. A trans-fatty acid-rich diet promotes liver tumorigenesis in HCV core gene transgenic mice. Carcinogenesis 2020, 41, 159–170. [Google Scholar] [CrossRef]
- Jia, F.P.; Diao, P.; Wang, X.J.; Hu, X.; Kimura, T.; Nakamuta, M.; Nakamura, I.; Shirotori, S.; Sato, Y.; Moriya, K.; et al. Dietary Restriction Suppresses Steatosis-Associated Hepatic Tumorigenesis in Hepatitis C Virus Core Gene Transgenic Mice. Liver Cancer 2020, 9, 529–548. [Google Scholar] [CrossRef]
- Zhang, Z.; Diao, P.; Zhang, X.G.; Nakajima, T.; Kimura, T.; Tanaka, N. Clinically Relevant Dose of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor alpha Modulator (SPPARM alpha), Lowers Serum Triglyceride Levels by Targeting Hepatic PPAR alpha in Mice. Biomedicines 2022, 10, 1667. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; Nagaya, T.; Horiuchi, A.; Tanaka, E. Clinical Features of Nonalcoholic Fatty Liver Disease Developed After Pancreaticoduodenectomy. Gastroenterology 2009, 136, A417–A418. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Kimura, T.; Kitabatake, H.; Fujimori, N.; Komatsu, M.; Horiuchi, A.; Yamaura, T.; Umemura, T.; Sano, K.; et al. Mechanism of the development of nonalcoholic steatohepatitis after pancreaticoduodenectomy. BBA Clin. 2015, 3, 168–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geier, A.; Dietrich, C.G.; Voigt, S.; Ananthanarayanan, M.; Lammert, F.; Schmitz, A.; Trauner, M.; Wasmuth, H.E.; Boraschi, D.; Balasubramaniyan, N.; et al. Cytokine-dependent regulation of hepatic organic anion transporter gene transactivators in mouse liver. Am. J. Physiol.-Gastrointest. Liver Physiol. 2005, 289, G831–G841. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Sakai, J.; Fujino, T.; Hattori, H.; Zenimaru, Y.; Suzuki, J.; Miyamori, I.; Yamamoto, T.T. The very low-density lipoprotein (VLDL) receptor: Characterization and functions as a peripheral lipoprotein receptor. J. Atheroscler. Thromb. 2004, 11, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Ito, T.; Zenimaru, Y.; Suzuki, J.; Miyamori, I.; Takahashi, M.; Ishida, T.; Hirata, K.; Yamamoto, T.T.; Iwasaki, T.; et al. Species differences of macrophage very low-density-lipoprotein (VLDL) receptor protein expression. Biochem. Biophys. Res. Commun. 2011, 407, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, H.Y.; Lee, H.S.; Jung, K.S.; Lee, M.H.; Jhee, J.H.; Kim, T.H.; Lee, J.E.; Kim, H.J.; Kim, B.S.; et al. Association between non-alcoholic fatty liver disease and coronary calcification depending on sex and obesity. Sci. Rep. 2020, 10, 1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of Cholesterol-Associated Steatohepatitis in the Development of NASH. Hepatol. Commun. 2022, 6, 12–35. [Google Scholar] [CrossRef]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2015, 1852, 1765–1778. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Landis, C.S.; Jin, G.Y.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated With Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef] [Green Version]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.L.; Rada, P.; Quesada-Lopez, T.; Escola-Gil, J.C.; Cedo, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPAR beta/delta and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.P.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased Hepatic Synthesis and Dysregulation of Cholesterol Metabolism Is Associated with the Severity of Nonalcoholic Fatty Liver Disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Kim, B.H.; Seo, H.S.; Lee, Y.J.; Kim, H.H.; Son, H.H.; Choi, M.H. Cholesterol-Induced Non-Alcoholic Fatty Liver Disease and Atherosclerosis Aggravated by Systemic Inflammation. PLoS ONE 2014, 9, e97841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, R.P.; Porat-Shliom, N. Liver Zonation—Revisiting Old Questions With New Technologies. Front. Physiol. 2021, 12, 732929. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, M.; Kuwabara, Y.; Ito, K.; Hojo, Y.; Arai, F.; Kamijima, K.; Takeiri, M.; Wang, X.; Diao, P.; Nakayama, J.; et al. Development of the Rabbit NASH Model Resembling Human NASH and Atherosclerosis. Biomedicines 2023, 11, 384. https://doi.org/10.3390/biomedicines11020384
Hayashi M, Kuwabara Y, Ito K, Hojo Y, Arai F, Kamijima K, Takeiri M, Wang X, Diao P, Nakayama J, et al. Development of the Rabbit NASH Model Resembling Human NASH and Atherosclerosis. Biomedicines. 2023; 11(2):384. https://doi.org/10.3390/biomedicines11020384
Chicago/Turabian StyleHayashi, Momoko, Yoshibumi Kuwabara, Kuniji Ito, Yoshiaki Hojo, Fumiaki Arai, Kazuki Kamijima, Masakazu Takeiri, Xiaojing Wang, Pan Diao, Jun Nakayama, and et al. 2023. "Development of the Rabbit NASH Model Resembling Human NASH and Atherosclerosis" Biomedicines 11, no. 2: 384. https://doi.org/10.3390/biomedicines11020384