
A Century of Progress on Wilson Disease and the Enduring Challenges of Genetics, Diagnosis, and Treatment

 , , , ,
, , , ,  , ,
, , 

Abstract
:1. Introduction
- Does a hyperimmune state with isolated elevation of autoantibodies predispose to liver inflammation?
- Is such hyperimmunity more frequent in Wilson disease?
- Is manifestation of liver disease frequently a multifactorial process?
- Can we identify a gold-standard for diagnosis of Wilson disease?
- Are currently available tests adequate to promptly distinguish Wilson disease from other chronic liver diseases?
- Wilson disease affects pediatric and young adults, but the patient population is aging with associated comorbidities. What is the best anti-copper treatment approach in elderly patients?
- When should we consider a change in anti-copper treatment strategy?
- How do we monitor any treatment transition? When can we consider a treatment change to be successful?
2. Specific Problems Related to Rare Diseases
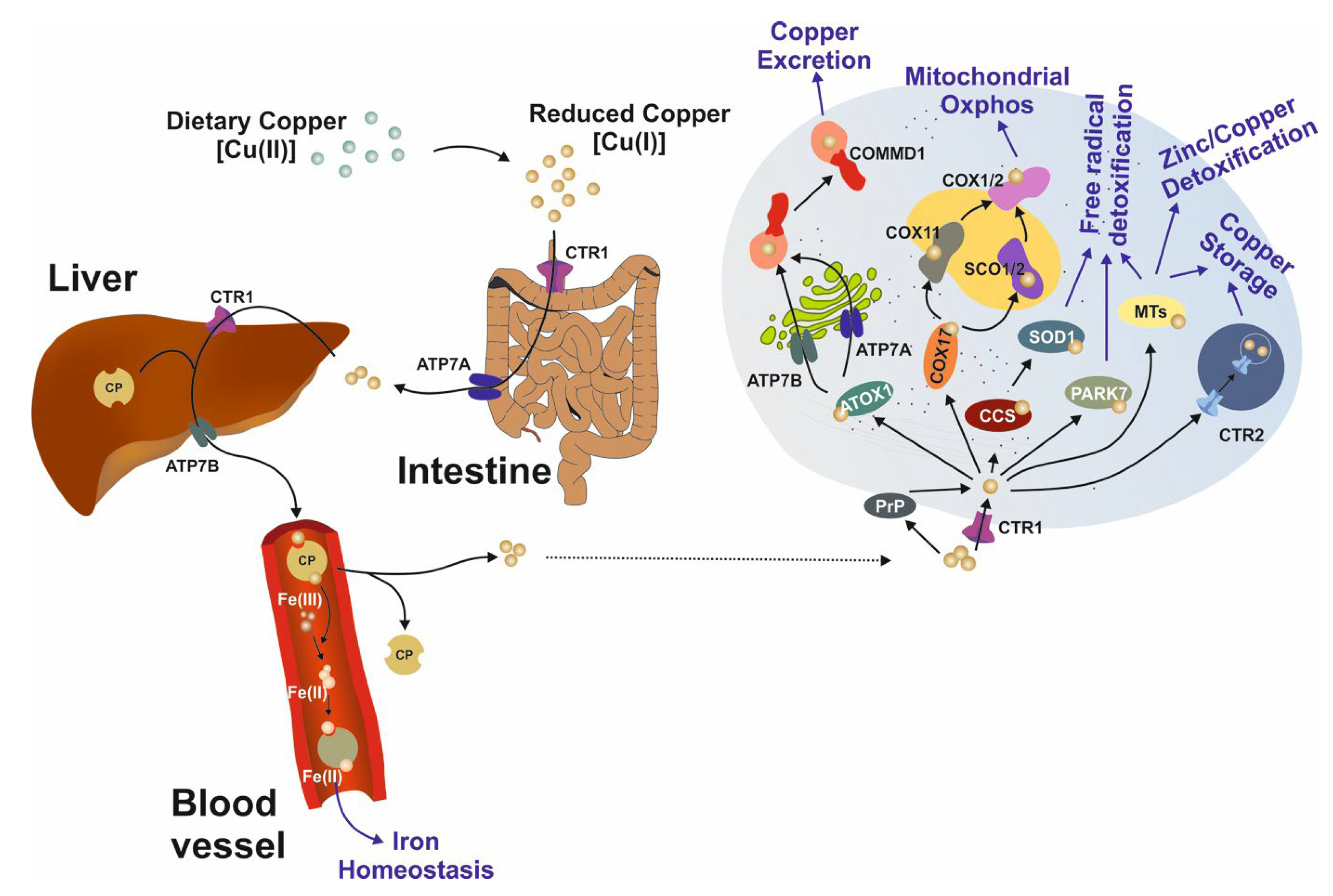
3. Intracellular Copper Transport
4. Genetics and Epidemiology
5. Hepatic and/or Neurological Presentation
- How can we identify WD patients who are at risk of neurological deterioration?
- Which strategies prevent, or attenuate the severity of, neurological deterioration after treatment initiation in WD patients?
- Should we do an MRI brain examination in patients with hepatic presentation, particularly in those with a normal neurological examination? We advocate this approach, because (i) MRI brain changes are detected in 40% of patients with the hepatic form of the disease [73], and (ii) the presence of changes in MRI is a predictor of early deterioration after starting treatment, especially with D-Penicillamine and lesions in thalami and pons [78].
- Should parents of WD index cases receive the same detailed screening (including DNA analysis) as siblings? We should carefully consider family history and include wider family members, not just siblings, in our investigations. As WD manifests mostly in young persons, detailed genetic studies of parents may not be required [80,81]. Nevertheless, carefully analyzing medical histories and basic laboratory tests are recommended. This is a rare case. WD was diagnosed in the parent of our index case; there is another case report of this type in the literature [82]. The pseudodominant (vertical) type of inheritance in WD is rarely diagnosed in our cohorts, being found in 4% of offspring [83].
- How can we better monitor the efficacy of copper chelation treatment during life?
- How are copper regulatory pathways altered in vulnerable organs, such as the liver and brain, in Wilson disease and do such changes explain variability in disease phenotype and in response to treatment?
- What modifier genes are related to variable hepatic copper levels and clinical presentation in WD patients? Can we use genetics of canine inbreeding to find modifier genes?
- What are the genetic predispositions to very rare copper-related disorders such as Indian childhood cirrhosis, endemic Tyrolean infantile cirrhosis and idiopathic copper toxicosis?
- When can we trust the advice of our health professionals? When should we seek a second opinion? How can we help our health professionals reach an accurate and early diagnosis?
- What can we do to raise awareness of the need for expert centers? How can we ensure that healthcare professionals connect to such centers and ask for WD expert advice when required?
- How can we empower young patients to take responsibility for the management of their disease—and support parents of young WD patients as they learn these skills?
- How can we educate healthcare professional such that they take our concerns seriously and are open to seeking expert advice when needed?
- How can we teach healthcare professionals to listen to our concerns and see us as people, not just patients or a problem to be solved?
- Complement: The unique experience or ’hands-on’ expertise of patients complements the knowledge and experience of other parties in health care, such as healthcare providers, scientists and policymakers.
- Meeting the needs of patients: Patient participation ensures that developments in research, policy, guideline development, and care innovation are better attuned to patients’ needs and expectations. It also provides insight into what patients and their relatives consider feasible and acceptable.
- Contributes to implementation: Patient participation in all phases of a project or research also contributes to its implementation—that something is done with the findings.
- Follow the decision-making processes and ensure a democratic process of patient representation;
- Provide the perspective of patients on all relevant aspects in the policy & organisational processes;
- Promote and encourage, where possible, a patient-centric approach in both delivery of clinical care, service improvement and strategic development and decision-making;
- Advocate for care that is patient-centred and respectful of patients’ rights and choices;
- Provide the patient perspective on the application of personal data rules, compliance of information consent, and the management of complaints;
- Ensure that processes to address all ethical issues and concerns for patients are in place, balancing patient and clinical needs appropriately;
- Advise on transparency in quality of care, safety standards, clinical outcomes, and treatment options;
- Monitor the performance of the results by reviewing quality indicators such as the clinical outcomes of diagnosis and treatment;
- Contribute to the development and dissemination of patient information, policy, good practice, care pathways, and guidelines;
- Advise to a general project website with a lay version (in the native language of several partners);
- Contribute to research, e.g., defining research areas important to patients and their families and disseminating research-related information in a lay version by regular articles in their quarterly magazine concerning relevant information about the project, on websites, and other social media platforms such as Facebook and Twitter;
- Share the information with their colleague patient organizations related to the subject;
- Inform their umbrella organizations by articles/reports and presentations during international meetings;
- Advise about providing a lay version of the final results of the project.
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilson, S.A.K. Degeneration lenticulaire progressive Maladie nerveuse familiale associee a la cirrhose de foi. Rev. Neurol. 1912, 23, 229–234. [Google Scholar]
- Wilson, S.A.K. Progressive lenticular degeneration: A familial nervous diseases associated with cirrhosis of the liver. Brain 1912, 34, 295–307. [Google Scholar] [CrossRef]
- Rumpel, A. Über das Wesen und die Bedeutung der Leberveränderungen und der Pigmentierungen bei den damit verbundenen Fällen von Pseudosklerose, zugleich ein Beitrag zur Lehre von der Pseudosklerose (Westphal-Strumpell). Dtsch. Z. Nervenheilkd. 1913, 49, 54–73. [Google Scholar] [CrossRef]
- Cumings, J.N. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain 1948, 71, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Walshe, J.M. The conquest of Wilson’s disease. Brain 2009, 132, 2289–2295. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Prim. 2018, 4, 21. [Google Scholar] [CrossRef]
- Frydman, M.; Bonne-Tamir, B.; Farrer, L.A.; Conneally, P.M.; Magazanik, A.; Ashbel, S.; Goldwitch, Z. Assignment of the gene for Wilson diseases to chromosome 13: Linkage to the esterase D locus. Proc. Natl. Acad. Sci. USA 1985, 82, 1819–1821. [Google Scholar] [CrossRef]
- Petrukhin, K.; Fischer, S.G.; Pirastu, M.; Tanzi, R.E.; Chernov, I.; Devoto, M.; Brustowicz, L.M.; Cayanis, E.; Vitale, E.; Russo, J.J.; et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat. Genet. 1993, 5, 338–343. [Google Scholar] [CrossRef]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson’s disease gene is a putative copper transporting P-type ATPase similar to Menkes’ gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Heiny, M.E.; Gitlin, J.D. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem. Biophys. Res. Commun. 1993, 197, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wakap, S.N.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Ophanet. The Portal for Rare Diseases and Orphan Drugs. Available online: www.orphanet.net (accessed on 15 May 2022).
- Lee, C.E.; Singleton, K.S.; Wallin, M.; Faundez, V. Rare Genetic Diseases: Nature’s Experiments on Human Development. iScience 2020, 23, 101123. [Google Scholar] [CrossRef] [PubMed]
- European Reference Network: Hepatological Diseases (ERN RARE-LIVER). Available online: https://rare-liver.eu (accessed on 15 May 2022).
- Gil-Bea, F.J.; Aldanondo, G.; Lasa-Fernández, H.; de Munain, A.L.; Vallejo-Illarramendi, A. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev. Mol. Med. 2017, 19, e7. [Google Scholar] [CrossRef]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Inesi, G. Molecular features of copper binding proteins involved in copper homeostasis. IUBMB Life 2017, 69, 211–217. [Google Scholar] [CrossRef]
- Reed, E.; Lutsenko, S.; Bandmann, O. Animal models of Wilson disease. J. Neurochem. 2018, 146, 356–373. [Google Scholar] [CrossRef]
- Pierson, H.; Yang, H.; Lutsenko, S. Copper Transport and Disease: What can we learn from organoids? Annu. Rev. Nutr. 2019, 39, 75–94. [Google Scholar] [CrossRef]
- Hartwig, C.; Zlatic, S.A.; Wallin, M.; Vrailas-Mortimer, A.; Fahrni, C.J.; Faundez, V. Trafficking mechanisms of P-type ATPase copper transporters. Curr. Opin. Cell Biol. 2019, 59, 24–33. [Google Scholar] [CrossRef]
- Polishchuk, R.S.; Polishchuk, E.V. From and to the Golgi—Defining the Wilson disease protein road map. FEBS Lett. 2019, 593, 2341–2350. [Google Scholar] [CrossRef]
- Stremmel, W.; Weiskirchen, R. Therapeutic strategies in Wilson disease: Pathophysiology and mode of action. Ann. Transl. Med. 2021, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S. Dynamic and cell-specific transport networks for intracellular copper ions. J. Cell Sci. 2021, 134, jcs240523. [Google Scholar] [CrossRef] [PubMed]
- Maung, M.T.; Carlson, A.; Olea-Flores, M.; Elkhadragy, L.; Schachtschneider, K.M.; Navarro-Tito, N.; Padilla-Benavides, T. The relationship and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 2021, 35, e21810. [Google Scholar] [CrossRef] [PubMed]
- Bitter, R.M.; Oh, S.; Deng, Z.; Rahman, S.; Hite, R.K.; Yuan, P. Structure of the Wilson disease copper transporter ATP7B. Sci. Adv. 2022, 8, eabl5508. [Google Scholar] [CrossRef] [PubMed]
- Zischka, H.; Lichtmannegger, J.; Schmitt, S.; Jägemann, N.; Schulz, S.; Wartini, D.; Jennen, L.; Rust, C.; Larochette, N.; Galluzzi, L.; et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J. Clin. Investig. 2011, 121, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Borchard, S.; Bork, F.; Rieder, T.; Eberhagen, C.; Popper, B.; Lichtmannegger, J.; Schmitt, S.; Adamski, J.; Klingenspor, M.; Weiss, K.H.; et al. The exceptional sensitivity of brain mitochondria to copper. Toxicol. Vitr. 2018, 51, 11–22. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dutta, S.; Majumdar, S.; Biswas, T.; Jaiswal, P.; Sengupta, M.; Bhattacharya, A.; Gangopadhyay, P.K.; Avdekar, A.; Das, S.K.; et al. Genetic defects in Indian Wilson disease patients and genotype-phenotype correlation. Park. Relat. Disord. 2014, 20, 75–81. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Coper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Parisi, S.; Polishchuk, E.V.; Allocca, S.; Ciano, M.; Musto, A.; Gallo, M.; Perone, L.; Ranucci, G.; Iorio, R.; Polishchuk, R.S.; et al. Characterization of the most frequent ATP7B mutations causing Wilson disease I hepatocytes from patient induced pluripotent stem cells. Sci. Rep. 2018, 8, 6247. [Google Scholar] [CrossRef]
- National Library of Medicine. ClinVar. Entry ATP7B Gene. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=atp7b%5Bgene%5D&redir=gene (accessed on 5 May 2022).
- The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ATP7B (accessed on 5 May 2022).
- Gao, J.; Brackley, S.; Mann, J.P. The global prevalence of Wilson disease from next-generation sequencing data. Genet. Med. 2019, 21, 1155–1163. [Google Scholar] [CrossRef]
- Leung, M.; Aronowotz, P.B.; Medici, V. The present and future challenges of Wilson’s disease diagnosis and treatment. Clin. Liver Dis. 2021, 17, 267–270. [Google Scholar] [CrossRef]
- Sanchez-Monteagudo, A.; Ripoles, E.; Berenguer, M.; Espinos, C. Wilson’s disease: Facing the challenge of diagnosing a rare disease. Biomedicines 2021, 9, 1100. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Villarreal, L.; Hernandez-Ortega, A.; Sanchez-Monteagudo, A.; Pena-Quintana, L.; Ramirez-Lorenzo, T.; Riano, M.; Moreno-Perez, R.; Monescillo, A.; Gonzalez_Santana, D.; Quinones, I.; et al. Wilson disease: Revision of diagnostic criteria in a clinical series with great genetic homogeneity. J. Gasteroenterol. 2021, 56, 78–89. [Google Scholar] [CrossRef]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A genetic study of Wilson’s disease in the United Kingdom. Brain 2013, 136, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Collet, C.; Laplanche, J.L.; Page, J.; Morel, H.; Woimasnt, F.; Poujois, A. High genetic carrier frequency of Wilson’s disease in France: Discrepancies with clinical prevalence. BMC Med. Genet. 2018, 19, 143. [Google Scholar] [CrossRef] [PubMed]
- Garcıa-Villarreal, L.; Daniels, S.; Shaw, S.H.; Cotton, D.; Galvin, M.; Geskes, J.; Bauer, P.; Sierra-Herandez, A.; Buckler, A.; Tugores, A. High prevalence of the very rare Wilson disease gene mutation Leu708Pro in the Island of Gran Canaria (Canary Islands, Spain): A genetic and clinical study. Hepatology 2000, 32, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Monteagudo, A.; lvarez-Sauco, M.; Sastre, I.; Martinez-Torres, I.; Lupo, V.; Berenguer, M.; Espinos, C. Genetics of Wilson disease and Wilson-like phenotype in a clinical series from eastern Spain. Clin. Genet. 2020, 97, 758–763. [Google Scholar] [CrossRef]
- Ferenci, P. Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: Impact on genetic testing. Hum. Genet. 2006, 120, 151–159. [Google Scholar] [CrossRef]
- Zappu, A.; Magli, O.; Lepori, M.B.; Dessi, V.; Diana, S.; Incollu, S.; Kanavakis, E.; Nicolaidou, P.; Manolaki, N.; Fretzayas, A.; et al. High incidence and allelic homogeneity of Wilson disease in 2 isolated populations: A prerequisite for efficient disease prevention programs. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 334–338. [Google Scholar] [CrossRef]
- Espinós, C.; Ferenci, P. Are the new genetic tools for diagnosis of Wilson disease helpful in clinical practice? JHEP Rep. 2020, 2, 100114. [Google Scholar] [CrossRef]
- Wallace, D.F.; Dooley, J.S. ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease. Hum. Genet. 2020, 139, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; Lasalle, J.M. Genetics and epigenetics factors of Wilson disease. Ann. Transl. Med. 2019, 7, S58. [Google Scholar] [CrossRef]
- Lalioti, V.; Tsubota, A.; Sandoval, I.V. Disorders in hepatic copper secretion: Wilson’s disease and pleomorphic syndromes. Semin. Liver Dis. 2017, 37, 175–188. [Google Scholar] [CrossRef]
- Mordaunt, C.E.; Kieffer, D.A.; Shibata, N.M.; Członkowska, A.; Litwin, T.; Weiss, K.H.; Zhu, Y.; Bowlus, C.L.; Sarkar, S.; Cooper, S.; et al. Epigenomic signatures in liver and blood of Wilson disease patients include hypermethylation of liver-specific enhancers. Epigenetics Chromatin 2019, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Moini, M.; To, U.; Schilsky, M.L. Recent advances in Wilson disease. Transl. Gastroenterol. Hepatol. 2021, 6, 21. [Google Scholar] [CrossRef]
- Stattermayer, A.F.; Traussnigg, S.; Dines, H.-P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease—Role of copper and PNPLA3 mutations. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef]
- Schiefermeier, M.; Kollegger, H.; Madl, C.; Polli, C.; Oder, W.; Kühn, H.-J.; Berr, F.; Ferenci, P. The impact of apolipoprotein E genotypes on age at onset of symptoms and phenotypic expression in Wilson’s disease. Brain 2000, 123, 585–590. [Google Scholar] [CrossRef]
- Medici, V.; Weiss, K.H. Genetic and environmental modifiers of Wilson disease. Handb. Clin. Neurol. 2017, 142, 35–41. [Google Scholar] [CrossRef]
- Simon, L.; Schaefer, M.; Reichert, J.; Stremmel, W. Analysis of the human atox1 homologue in Wilson patients. World J. Gastroenterol. 2008, 14, 2383–2387. [Google Scholar] [CrossRef]
- Bost, M.; Piguit-Lacroix, G.; Parant, F.; Wilson, C.M.R. Molecular analysis of Wilson patients: Direct sequencing and MLPA analysis in the ATP7B gene and Atox1 and COMMD1 gene analysis. J. Trace Elem. Med. Biol. 2012, 26, 97–101. [Google Scholar] [CrossRef]
- Kumari, N.; Kumar, A.; Pal, A.; Thapa, B.R.; Modi, M.; Prasad, R. In-silico analysis of novel p.(Gly14Ser) variant of ATOX1 gene: Plausible role in modulating ATOX1-ATP7B interaction. Mol. Biol. Rep. 2019, 46, 3307–3313. [Google Scholar] [CrossRef] [PubMed]
- Zarina, A.; Tolmane, I.; Krumina, Z.; Tutane, A.I.; Gailite, L. Association of variants in the CP, ATOX1, and COMMD1 genes with Wilson disease symptoms in Latvia. Balk. J. Med. Genet. 2019, 21, 37–42. [Google Scholar] [CrossRef] [PubMed]
- van de Sluis, B.; Rothuizen, J.; Pearson, P.L.; van Oost, B.; Wijmenga, C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum. Mol. Genet. 2002, 11, 165–173. [Google Scholar] [CrossRef]
- Stuehler, B.; Reichert, J.; Stremmel, W.; Schaefer, M. Analysis of the human homologue of the canine copper toxicosis gene MURR1. J. Mol. Med. 2004, 82, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Merle, U.; Schaefer, M.; Ferenci, P.; Fullekrug, J.; Stremmel, W. Copper toxicosis gene MURR1 is not changed in Wilson disease patients with normal blood ceruloplasmin levels. World J. Gastroenterol. 2006, 12, 2239–2242. [Google Scholar] [CrossRef]
- Gupta, A.; Chattopadhyay, I.; Mukherjee, S.; Sengupta, M.; Das, S.K.; Ray, K. A novel COMMD1 mutation Thr174Met associated with elevated urinary copper and signs of enhanced apoptotic cell death in a Wilson Disease patient. Behav. Brain Funct. 2010, 6, 33. [Google Scholar] [CrossRef]
- Weiss, K.H.; Runz, H.; Noe, B.; Gotthardt, D.N.; Merle, U.; Ferenci, P.; Stremmel, W.; Fuellekrug, J. Genetic analysis of BIRC4/XIAP as a putative modifier gene of Wilson disease. J. Inherit. Metab. Dis. 2010, 33, S233–S240. [Google Scholar] [CrossRef]
- Hafkemeyer, P.; Schupp, M.; Storch, M.; Gertok, W.; Hausinger, D. Excessive iron storage in a patient with Wilson’s disease. J. Mol. Med. 1994, 72, 134–136. [Google Scholar] [CrossRef]
- Walshe, J.M.; Cox, D.W. Effect of treatment of Wilson’s disease on natural history of haemochromatosis. Lancet 1998, 352, 112–113. [Google Scholar] [CrossRef]
- Sorbello, O.; Sini, M.; Civolani, A.; Demelia, L. HFE gene mutations and Wilson’s disease in Sardinia. Dig. Liver Dis. 2010, 42, 216–219. [Google Scholar] [CrossRef]
- Pfeffenberger, J.; Gotthardt, D.N.; Herrmann, T.; Seesle, J.; Merle, U.; Schirmacher, P.; Stremmel, W.; Weiss, K.H. Iron metabolism and the role of HFE gene polymorphism in Wilson disease. Liver Int. 2012, 32, 165–170. [Google Scholar] [CrossRef]
- Przybylkowski, A.; Gromadzka, G.; Czlonkowska, A. Polymorphism of metal transporter genes DMT1 and ATP7A in Wilson’s disease. J. Trace Elem. Med. Biol. 2014, 28, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Sibani, S.; Christensen, B.; O’Farrall, E.; Saadi, I.; Hiou-Tim, F.; Rosenblatt, D.S.; Rozen, R. Characterization of six novel mutations in the methylenetetrahydrofilate reductase (MTHFR) gene in patients with homocystinuria. Hum. Mutat. 2000, 15, 280–287. [Google Scholar] [CrossRef]
- Gromadzka, G.; Rudnicka, M.; Chabik, G.; Przybylkowski, A.; Czlonkowska, A. Genetic variability in the methylenetetrahydroflate reductase gene (MTHFR) affects clinical expression of Wilson’s disease. J. Hepatol. 2011, 55, 913–919. [Google Scholar] [CrossRef]
- Poldervaart, J.; Favier, R.P.; Penning, L.C.; van den Ingh, T.S.; Rothuizen, J. Primary hepatitis in dogs: A retrospective review (2002–2006). J. Vet. Intern. Med. 2009, 23, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Fieten, H.; Penning, L.C.; Leegwater, P.A.J.; Rothuizen, J. New canine models of copper toxicosis: Diagnosis, treatment, and genetics. Ann. N. Y. Acad. Sci. 2014, 1314, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Fieten, H.; Gill, Y.; Martin, A.J.; Concilli, M.; Dirksen, K.; van Steenbeek, F.G.; Spee, B.; van den Ingh, T.S.; Martens, E.C.; Festa, P.; et al. The Menkes and Wilson disease genes counteract in copper toxicosis in Labrador retrievers: A new canine model for copper-metabolism disorders. Dis. Model Mech. 2016, 9, 25–38. [Google Scholar] [CrossRef]
- Reuner, U.; Dinger, J. Pregnancy and Wilson disease: Management and outcome of mother and newborns-experiences of a perinatal centre. Ann. Transl. Med. 2019, 7 (Suppl. 2), S56. [Google Scholar] [CrossRef]
- Litwin, T.; Gromadzka, G.; Czlonkowska, A.; Golebiowski, M.; Poniatowska, R. The effect of gender on brain MRI pathology in Wilson’s disease. Metab. Brain Dis. 2013, 28, 69–75. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [CrossRef]
- Medici, V.; Huster, D. Animal models of Wilson disease. Handb. Clin. Neurol. 2017, 142, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Huster, D. Chapter 13—Animal models for Wilson disease. In Clinical and Translational Perspectives on Wilson Disease; Kerkar, N., Roberts, E.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 127–139. [Google Scholar] [CrossRef]
- Dusek, P.; Skoloudik, D.; Maskova, J.; Huelnhagen, T.; Bruha, R.; Zahorakova, D.; Niendorf, T.; Ruzicka, E.; Schneider, S.A.; Wuerfel, J. Brain iron accumulation in Wilson’s disease: A longitudinal imaging case study during anticopper treatment using 7.0T MRI and transcranial sonography. J. Magn. Reson Imaging 2018, 47, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.; Dzieżyc, K.; Karliński, M.; Chabik, G.; Czepiel, W.; Członkowska, A. Early neurological worsening in patients with Wilson’s disease. J. Neurol. Sci. 2015, 355, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Członkowska, A.; Rodo, M.; Wierzchowska-Ciok, A.; Smolinski, L.; Litwin, T. Accuracy of the radioactive copper incorporation test in the diagnosis of Wilson disease. Liver Int. 2018, 38, 1860–1866. [Google Scholar] [CrossRef]
- Ferenci, P.; Członkowska, A.; Merle, U.; Ferenc, S.; Gromadzka, G.; Yurdaydin, C.; Vogel, W.; Bruha, R.; Schmidt, H.T.; Stremmel, W. Late-onset Wilson’s disease. Gastroenterology 2007, 132, 1294–1298. [Google Scholar] [CrossRef]
- Członkowska, A.; Rodo, M.; Gromadzka, G. Late onset Wilson’s disease: Therapeutic implications. Mov. Disord. 2008, 23, 896–898. [Google Scholar] [CrossRef]
- Brunet, A.S.; Marotte, S.; Guillaud, O.; Lachaux, A. Familial screening in Wilson’s disease: Think at the previous generation. J. Hepatol. 2012, 57, 1394–1395. [Google Scholar] [CrossRef]
- Dzieżyc, K.; Litwin, T.; Chabik, G.; Gramza, K.; Członkowska, A. Families with Wilson’s disease in subsequent generations: Clinical and genetic analysis. Mov. Disord. 2014, 29, 1828–1832. [Google Scholar] [CrossRef]
- Genoud, S.; Senior, A.M.; Hare, D.J.; Double, K.L. Meta-analysis of copper and iron in Parkinson’s disease brain and biofluids. Mov. Disord. 2020, 35, 662–671. [Google Scholar] [CrossRef]
- Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Topic | Question |
|---|---|
| Genetics | How can we best use DNA sequencing to improve WD diagnostics? |
| Can we discover additional modifier genes? | |
| Diagnosis | How can we improve monitoring the effects of copper chelation? |
| How can we improve specific copper measurements? | |
| How can we improve diagnostics in neurological WD? | |
| Is the current gold standard for WD diagnosis sufficient to discriminate WD from other hepatic diseases? | |
| What is the role for autoimmune antibodies in WD? | |
| Treatment | Should treatment and treatment monitoring be individualized based on patient age? |
| What is the best approach for monitoring the efficacy of treatment for neurological WD? | |
| Disease modeling | How are copper-regulatory pathways altered in WD? |
| How relevant are organoid models? | |
| Patient perspective | What is the best advice from healthcare professionals for patients? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penning, L.C.; Berenguer, M.; Czlonkowska, A.; Double, K.L.; Dusek, P.; Espinós, C.; Lutsenko, S.; Medici, V.; Papenthin, W.; Stremmel, W.; et al. A Century of Progress on Wilson Disease and the Enduring Challenges of Genetics, Diagnosis, and Treatment. Biomedicines 2023, 11, 420. https://doi.org/10.3390/biomedicines11020420
Penning LC, Berenguer M, Czlonkowska A, Double KL, Dusek P, Espinós C, Lutsenko S, Medici V, Papenthin W, Stremmel W, et al. A Century of Progress on Wilson Disease and the Enduring Challenges of Genetics, Diagnosis, and Treatment. Biomedicines. 2023; 11(2):420. https://doi.org/10.3390/biomedicines11020420
Chicago/Turabian StylePenning, Louis C., Marina Berenguer, Anna Czlonkowska, Kay L. Double, Petr Dusek, Carmen Espinós, Svetlana Lutsenko, Valentina Medici, Wiebke Papenthin, Wolfgang Stremmel, and et al. 2023. "A Century of Progress on Wilson Disease and the Enduring Challenges of Genetics, Diagnosis, and Treatment" Biomedicines 11, no. 2: 420. https://doi.org/10.3390/biomedicines11020420
APA StylePenning, L. C., Berenguer, M., Czlonkowska, A., Double, K. L., Dusek, P., Espinós, C., Lutsenko, S., Medici, V., Papenthin, W., Stremmel, W., Willemse, J., & Weiskirchen, R. (2023). A Century of Progress on Wilson Disease and the Enduring Challenges of Genetics, Diagnosis, and Treatment. Biomedicines, 11(2), 420. https://doi.org/10.3390/biomedicines11020420