
Proprotein Convertase Subtilisin/Kexin 9 as a Modifier of Lipid Metabolism in Atherosclerosis
 ,
,  ,
,
Abstract
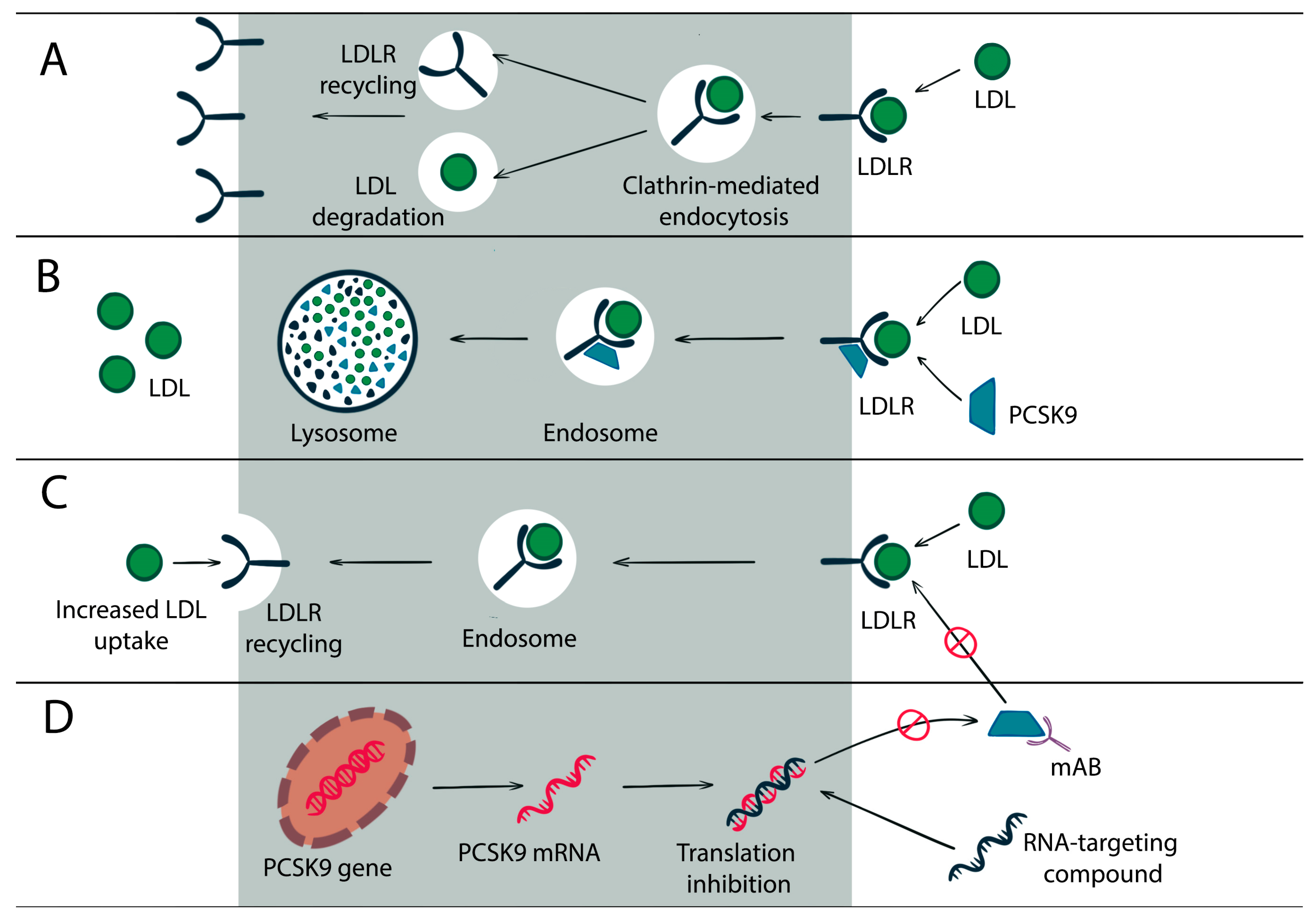
:1. PCSK9 Gene and Protein
2. PCSK9 Role in Lipid Homeostasis
3. PCSK9 in Vascular Inflammation
4. PCSK9 in Plaque Formation
5. Current PCSK9 Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Maligłówka, M.; Kosowski, M.; Hachuła, M.; Cyrnek, M.; Bułdak, Ł.; Basiak, M.; Bołdys, A.; Machnik, G.; Bułdak, R.J.; Okopień, B. Insight into the Evolving Role of PCSK9. Metabolites 2022, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Żak, J.; Malinowski, B.; Popek, G.; Grześk, G. PCSK9 signaling pathways and their potential importance in clinical practice. EPMA J. 2017, 8, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, P.F.; Byun, J.H.; Platko, K.; Saliba, P.; Sguazzin, M.; MacDonald, M.E.; Paré, G.; Steinberg, G.R.; Janssen, L.J.; Igdoura, S.A.; et al. Caffeine blocks SREBP2-induced hepatic PCSK9 expression to enhance LDLR-mediated cholesterol clearance. Nat. Commun. 2022, 13, 770. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Dong, B.; Cao, A.; Li, H.; Liu, J. Delineation of molecular pathways that regulate hepatic PCSK9 and LDL receptor expression during fasting in normolipidemic hamsters. Atherosclerosis 2012, 224, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Chen, Y.; Hu, W.; Li, X.; Yang, X.; Zhou, X.; Yin, Z.; Kong, D.; Yao, Z.; Hajjar, D.P.; et al. Peroxisome Proliferator-activated receptor γ activation by ligands and dephosphorylation induces proprotein convertase subtilisin kexin type 9 and low density lipoprotein receptor expression. J. Biol. Chem. 2012, 287, 23667–23677. [Google Scholar] [CrossRef]
- Xia, X.D.; Peng, Z.S.; Gu, H.M.; Wang, M.; Wang, G.Q.; Zhang, D.W. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef]
- Glerup, S.; Schulz, R.; Laufs, U.; Schlüter, K.D. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease. Basic Res. Cardiol. 2017, 112, 32. [Google Scholar] [CrossRef]
- Poirier, S.; Mayer, G.; Benjannet, S.; Bergeron, E.; Marcinkiewicz, J.; Nassoury, N.; Mayer, H.; Nimpf, J.; Prat, A.; Seidah, N.G. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 2008, 283, 2363–2372. [Google Scholar] [CrossRef]
- Papotti, B.; Adorni, M.P.; Marchi, C.; Zimetti, F.; Ronda, N.; Panighel, G.; Lupo, M.G.; Vilella, A.; Giuliani, D.; Ferri, N.; et al. PCSK9 Affects Astrocyte Cholesterol Metabolism and Reduces Neuron Cholesterol Supplying In Vitro: Potential Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12192. [Google Scholar] [CrossRef]
- Barale, C.; Melchionda, E.; Morotti, A.; Russo, I. PCSK9 Biology and Its Role in Atherothrombosis. Int. J. Mol. Sci. 2021, 22, 5880. [Google Scholar] [CrossRef]
- Lagace, T.A. PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef]
- Seidah, N.G.; Garçon, D. Expanding Biology of PCSK9: Roles in Atherosclerosis and Beyond. Curr. Atheroscler. Rep. 2022, 24, 821–830. [Google Scholar] [CrossRef]
- Pelletier, R.M.; Layeghkhavidaki, H.; Seidah, N.G.; Prat, A.; Vitale, M.L. PCSK9 Contributes to the Cholesterol, Glucose, and Insulin2 Homeostasis in Seminiferous Tubules and Maintenance of Immunotolerance in Testis. Front. Cell Dev. Biol. 2022, 10, 889972. [Google Scholar] [CrossRef]
- Jang, H.D.; Lee, S.E.; Yang, J.; Lee, H.C.; Shin, D.; Lee, H.; Lee, J.; Jin, S.; Kim, S.; Lee, S.J.; et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur. Heart J. 2020, 41, 239–252. [Google Scholar] [CrossRef]
- Mbikay, M.; Mayne, J.; Sirois, F.; Fedoryak, O.; Raymond, A.; Noad, J.; Chrétien, M. Mice Fed a High-Cholesterol Diet Supplemented with Quercetin-3-Glucoside Show Attenuated Hyperlipidemia and Hyperinsulinemia Associated with Differential Regulation of PCSK9 and LDLR in their Liver and Pancreas. Mol. Nutr. Food Res. 2018, 62, e1700729. [Google Scholar] [CrossRef]
- Momtazi, A.A.; Banach, M.; Pirro, M.; Katsiki, N.; Sahebkar, A. Regulation of PCSK9 by nutraceuticals. Pharmacol. Res. 2017, 120, 157–169. [Google Scholar] [CrossRef]
- Alannan, M.; Seidah, N.G.; Merched, A.J. PCSK9 in Liver Cancers at the Crossroads between Lipid Metabolism and Immunity. Cells 2022, 11, 4132. [Google Scholar] [CrossRef]
- Faraj, M. LDL, LDL receptors, and PCSK9 as modulators of the risk for type 2 diabetes: A focus on white adipose tissue. J. Biomed. Res. 2020, 34, 251–259. [Google Scholar] [CrossRef]
- Grewal, T.; Buechler, C. Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond. Int. J. Mol. Sci. 2022, 23, 1070. [Google Scholar] [CrossRef]
- Rashid, S.; Tavori, H.; Brown, P.E.; Linton, M.F.; He, J.; Giunzioni, I.; Fazio, S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation 2014, 130, 431–441, Erratum in Circulation 2015, 131, e429. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Lagace, T.A.; McNutt, M.C.; Horton, J.D.; Deisenhofer, J. Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. USA 2008, 105, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Moradi, A.; Maleki, M.; Ghaemmaghami, Z.; Khajali, Z.; Noohi, F.; Moghadam, M.H.; Kalyinia, S.; Mowla, S.J.; Seidah, N.G.; Malakootian, M. Mutational Spectrum of LDLR and PCSK9 Genes Identified in Iranian Patients with Premature Coronary Artery Disease and Familial Hypercholesterolemia. Front. Genet. 2021, 12, 625959. [Google Scholar] [CrossRef] [PubMed]
- De Castro-Orós, I.; Pocoví, M.; Civeira, F. The genetic basis of familial hypercholesterolemia: Inheritance, linkage, and mutations. Appl. Clin. Genet. 2010, 3, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G. The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. Glob. Cardiol. Sci. Pract. 2017, 2017, e201702. [Google Scholar] [CrossRef]
- Uribe, K.B.; Chemello, K.; Larrea-Sebal, A.; Benito-Vicente, A.; Galicia-Garcia, U.; Bourane, S.; Jaafar, A.K.; Lambert, G.; Martín, C. A Systematic Approach to Assess the Activity and Classification of PCSK9 Variants. Int. J. Mol. Sci. 2021, 22, 13602. [Google Scholar] [CrossRef]
- Tavori, H.; Giunzioni, I.; Predazzi, I.M.; Plubell, D.; Shivinsky, A.; Miles, J.; Devay, R.M.; Liang, H.; Rashid, S.; Linton, M.F.; et al. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc. Res. 2016, 110, 268–278. [Google Scholar] [CrossRef]
- Guo, Q.; Feng, X.; Zhou, Y. PCSK9 Variants in Familial Hypercholesterolemia: A Comprehensive Synopsis. Front. Genet. 2020, 11, 1020. [Google Scholar] [CrossRef]
- Taghizadeh Jazdani, S.; Shahbazian, H.B.; Cheraghian, B.; Jalali, M.T.; Mohammadtaghvaei, N. Association between the rs615563 variant of PCSK9 gene and circulating lipids and Type 2 diabetes. BMC Res. Notes 2021, 14, 309. [Google Scholar] [CrossRef]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular biology of PCSK9: Its role in LDL metabolism. Trends Biochem. Sci. 2007, 32, 71–77. [Google Scholar] [CrossRef]
- Alannan, M.; Fatrouni, H.; Trézéguet, V.; Dittrich-Domergue, F.; Moreau, P.; Siegfried, G.; Liet, B.; Khatib, A.M.; Grosset, C.F.; Badran, B.; et al. Targeting PCSK9 in Liver Cancer Cells Triggers Metabolic Exhaustion and Cell Death by Ferroptosis. Cells 2022, 12, 62. [Google Scholar] [CrossRef]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Tabas, I.; Lichtman, A.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2017, 47, 621–634. [Google Scholar] [CrossRef]
- Ricci, C.; Ruscica, M.; Camera, M.; Rossetti, L.; Macchi, C.; Colciago, A.; Zanotti, I.; Lupo, M.G.; Adorni, M.P.; Cicero, A.F.G.; et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci. Rep. 2018, 8, 2267. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Tang, Z.H.; Peng, J.; Ren, Z.; Yang, J.; Li, T.T.; Li, T.H.; Wang, Z.; Wei, D.H.; Liu, L.S.; Zheng, X.L.; et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis 2017, 262, 113–122. [Google Scholar] [CrossRef]
- Lee, S.; Bartlett, B.; Dwivedi, G. Adaptive Immune Responses in Human Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 9322. [Google Scholar] [CrossRef]
- Yurtseven, E.; Ural, D.; Baysal, K.; Tokgözoğlu, L. An Update on the Role of PCSK9 in Atherosclerosis. J. Atheroscler. Thromb. 2020, 27, 909–918. [Google Scholar] [CrossRef]
- Bäck, M.; Yurdagul AJr Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Theus, S.; Deng, X.; Fan, Y.; Zhou, S.; Mehta, J.L. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. 2018, 114, 1145–1153, Erratum in Cardiovasc. Res. 2022, 118, 1849. [Google Scholar] [CrossRef]
- Luquero, A.; Badimon, L.; Borrell-Pages, M. PCSK9 Functions in Atherosclerosis Are Not Limited to Plasmatic LDL-Cholesterol Regulation. Front. Cardiovasc. Med. 2021, 8, 639727. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Jaafari, M.R.; Badiee, A.; Banach, M.; Sahebkar, A. Therapeutic effect of nanoliposomal PCSK9 vaccine in a mouse model of atherosclerosis. BMC Med. 2019, 17, 223. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, X.; Liu, S.; Zhou, S.; Kore, R.A.; Mu, S.; Deng, X.; Fan, Y.; Mehta, J.L. NLRP3 inflammasome via IL-1β regulates PCSK9 secretion. Theranostics 2020, 10, 7100–7110, Erratum in Theranostics 2022, 12, 418. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Nissen, S.E.; Prati, F.; Windecker, S.; Kataoka, Y.; Puri, R.; Hucko, T.; Kassahun, H.; Liao, J.; Somaratne, R.; et al. Assessing the impact of PCSK9 inhibition on coronary plaque phenotype with optical coherence tomography: Rationale and design of the randomized, placebo-controlled HUYGENS study. Cardiovasc. Diagn. Ther. 2021, 11, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhu, C.G.; Guo, Y.L.; Xu, R.X.; Zhang, Y.; Sun, J.; Li, J.J. The relationship between the plasma PCSK9 levels and platelet indices in patients with stable coronary artery disease. J. Atheroscler. Thromb. 2015, 22, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Pastori, D.; Nocella, C.; Farcomeni, A.; Bartimoccia, S.; Santulli, M.; Vasaturo, F.; Carnevale, R.; Menichelli, D.; Violi, F.; Pignatelli, P.; et al. Relationship of PCSK9 and Urinary Thromboxane Excretion to Cardiovascular Events in Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1455–1462. [Google Scholar] [CrossRef]
- Puteri, M.U.; Azmi, N.U.; Kato, M.; Saputri, F.C. PCSK9 Promotes Cardiovascular Diseases: Recent Evidence about Its Association with Platelet Activation-Induced Myocardial Infarction. Life 2022, 12, 190. [Google Scholar] [CrossRef]
- Krähenbühl, S.; Pavik-Mezzour, I.; von Eckardstein, A. Unmet Needs in LDL-C Lowering: When Statins Won’t Do! Drugs 2016, 76, 1175–1190. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Skavdis, A.; Sourlas, A.; Papakonstantinou, E.J.; Peña Genao, E.; Echavarria Uceta, R.; Guzman, E. Safety and Tolerability of PCSK9 Inhibitors: Current Insights. Clin. Pharmacol. 2020, 12, 191–202. [Google Scholar] [CrossRef]
- Nozue, T. Lipid Lowering Therapy and Circulating PCSK9 Concentration. J. Atheroscler. Thromb. 2017, 24, 895–907. [Google Scholar] [CrossRef]
- Barkas, F.; Elisaf, M.; Klouras, E.; Dimitriou, T.; Tentolouris, N.; Liberopoulos, E. Statin escape phenomenon: Fact or fiction? World J. Exp. Med. 2017, 7, 25–30. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Pedersen, T.R.; Park, J.G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: A prespecified secondary analysis of the FOURIER trial. Lancet 2017, 390, 1962–1971. [Google Scholar] [CrossRef]
- Ray, K.K.; Colhoun, H.M.; Szarek, M.; Baccara-Dinet, M.; Bhatt, D.L.; Bittner, V.A.; Budaj, A.J.; Diaz, R.; Goodman, S.G.; Hanotin, C.; et al. Effects of alirocumab on cardiovascular and metabolic outcomes after acute coronary syndrome in patients with or without diabetes: A prespecified analysis of the ODYSSEY OUTCOMES randomised controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 618–628, Erratum in Lancet Diabetes Endocrinol. 2019, 7, e21. [Google Scholar] [CrossRef]
- Koenig, W.; Conde, L.G.; Landmesser, U.; Leiter, L.A.; Ray, K.K.; Schwartz, G.G.; Wright, R.S.; Han, J.; Raal, F.J. Efficacy and Safety of Inclisiran in Patients with Polyvascular Disease: Pooled, Post Hoc Analysis of the ORION-9, ORION-10, and ORION-11 Phase 3 Randomized Controlled Trials. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef]
- Cordero, A.; Santos-Gallego, C.G.; Fácila, L.; Rodríguez-Mañero, M.; Bertomeu-González, V.; Castellano, J.M.; Seijas-Amigo, J.; Núñez, J.; Zuazola, P.; González-Juanatey, J.R.; et al. Estimation of the major cardiovascular events prevention with Inclisiran. Atherosclerosis 2020, 313, 76–80. [Google Scholar] [CrossRef]
- Nguyen, M.A.; Kosenko, T.; Lagace, T.A. Internalized PCSK9 dissociates from recycling LDL receptors in PCSK9-resistant SV-589 fibroblasts. J. Lipid Res. 2014, 55, 266–275. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef]
- Cicero, A.F.; Tartagni, E.; Ertek, S. Efficacy and safety profile of evolocumab (AMG145), an injectable inhibitor of the proprotein convertase subtilisin/kexin type 9: The available clinical evidence. Expert Opin. Biol. Ther. 2014, 14, 863–868. [Google Scholar] [CrossRef]
- Djebli, N.; Martinez, J.M.; Lohan, L.; Khier, S.; Brunet, A.; Hurbin, F.; Fabre, D. Target-Mediated Drug Disposition Population Pharmacokinetics Model of Alirocumab in Healthy Volunteers and Patients: Pooled Analysis of Randomized Phase I/II/III Studies. Clin. Pharmacokinet. 2017, 56, 1155–1171. [Google Scholar] [CrossRef]
- Martinez, J.M.; Brunet, A.; Hurbin, F.; DiCioccio, A.T.; Rauch, C.; Fabre, D. Population Pharmacokinetic Analysis of Alirocumab in Healthy Volunteers or Hypercholesterolemic Subjects Using a Michaelis-Menten Approximation of a Target-Mediated Drug Disposition Model-Support for a Biologics License Application Submission: Part I. Clin. Pharmacokinet. 2019, 58, 101–113. [Google Scholar] [CrossRef]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Blom, D.; Seidah, N.G.; Honarpour, N.; Lira, A.; Xue, A.; Chiruvolu, P.; et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: An analysis of 10 clinical trials and the LDL receptor’s role. J. Lipid Res. 2016, 57, 1086–1096. [Google Scholar] [CrossRef] [Green Version]
- Gaudet, D.; Kereiakes, D.J.; McKenney, J.M.; Roth, E.M.; Hanotin, C.; Gipe, D.; Du, Y.; Ferrand, A.C.; Ginsberg, H.N.; Stein, E.A. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am. J. Cardiol. 2014, 114, 711–715. [Google Scholar] [CrossRef] [PubMed]
- McKenney, J.M.; Koren, M.J.; Kereiakes, D.J.; Hanotin, C.; Ferrand, A.C.; Stein, E.A. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J. Am. Coll. Cardiol. 2012, 59, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.; Turner, T.; Visseren, F.L.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wen, D.; Chen, Y.; Ma, L.; You, C. PCSK9 inhibitors for secondary prevention in patients with cardiovascular diseases: A bayesian network meta-analysis. Cardiovasc. Diabetol. 2022, 21, 107. [Google Scholar] [CrossRef]
- Kaddoura, R.; Orabi, B.; Salam, A.M. Efficacy and safety of PCSK9 monoclonal antibodies: An evidence-based review and update. J. Drug Assess. 2020, 9, 129–144. [Google Scholar] [CrossRef]
- Hines, D.M.; Rane, P.; Patel, J.; Harrison, D.J.; Wade, R.L. Treatment patterns and patient characteristics among early initiators of PCSK9 inhibitors. Vasc. Health Risk Manag. 2018, 14, 409–418. [Google Scholar] [CrossRef]
- Piccinni, C.; Antonazzo, I.C.; Maggioni, A.P.; Pedrini, A.; Calabria, S.; Ronconi, G.; Dondi, L.; Martini, N.; Roberto, G.; Sampietro, T.; et al. PCSK9 Inhibitors’ New Users: Analysis of Prescription Patterns and Patients’ Characteristics from an Italian Real-world Study. Clin. Drug Investig. 2020, 40, 173–181. [Google Scholar] [CrossRef]
- German, C.A.; Shapiro, M.D. Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs 2020, 34, 1–9. [Google Scholar] [CrossRef]
- Pirillo, A.; Catapano, A.L. Inclisiran: How Widely and When Should We Use It? Curr. Atheroscler. Rep. 2022, 24, 803–811. [Google Scholar] [CrossRef]
- Merćep, I.; Friščić, N.; Strikić, D.; Reiner, Ž. Advantages and Disadvantages of Inclisiran: A Small Interfering Ribonucleic Acid Molecule Targeting PCSK9-A Narrative Review. Cardiovasc. Ther. 2022, 2022, 8129513. [Google Scholar] [CrossRef]
- Nishikido, T.; Ray, K.K. Non-antibody Approaches to Proprotein Convertase Subtilisin Kexin 9 Inhibition: siRNA, Antisense Oligonucleotides, Adnectins, Vaccination, and New Attempts at Small-Molecule Inhibitors Based on New Discoveries. Front. Cardiovasc. Med. 2019, 5, 199. [Google Scholar] [CrossRef]
- Brandts, J.; Ray, K.K. Clinical implications and outcomes of the ORION Phase III trials. Future Cardiol. 2021, 17, 769–777. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Cupido, A.J.; Laufs, U. Gene Therapy Targeting PCSK9. Metabolites 2022, 12, 70. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; Döring, Y.; van der Vorst, E.P.C. PCSK9: A Multi-Faceted Protein That Is Involved in Cardiovascular Biology. Biomedicines 2021, 9, 793. [Google Scholar] [CrossRef]
- Liu, C.; Chen, J.; Chen, H.; Zhang, T.; He, D.; Luo, Q.; Chi, J.; Hong, Z.; Liao, Y.; Zhang, S.; et al. PCSK9 Inhibition: From Current Advances to Evolving Future. Cells 2022, 11, 2972. [Google Scholar] [CrossRef]
- Basiak, M.; Hachula, M.; Kosowski, M.; Okopien, B. Effect of PCSK9 Inhibitors on Hemostasis in Patients with Isolated Hypercholesterolemia. J. Clin. Med. 2022, 11, 2542. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Trial | Drug | Dosage | Subjects | LDL-C | Reference |
|---|---|---|---|---|---|
| FOURIER | Evolocumab (with statins) | 140 mg biweekly or 420 mg monthly | 27,564 ASCVD patients | Reduction by 60% | [51] |
| ODYSSEY OUTCOMES | Alirocumab (with statins) | 75 to 150 mg biweekly | 18,924 ASCVD patients | Reduction by more than 50% | [52] |
| ORION 9 | Inclisiran | 284 mg on day 1, day 90, and 6-monthly thereafter | 482 heterozygous familial hypercholesterolaemia patients | Reduction by 53.8% | [53] |
| ORION 10 | Inclisiran | 284 mg on day 1, day 90, and 6-monthly thereafter | 1561 ASCVD and elevated LDL-C patients | Reduction by 47.2% | [53] |
| ORION 11 | Inclisiran | 284 mg on day 1, day 90, and 6-monthly thereafter | 1617 ASCVD or risk equivalents and elevated LDL-C patients | Reduction by 47.9% | [53] |
| ORION 4 | Inclisiran | 300 milligram (mg) inclisiran sodium for injection administered by SC injection on day 1 and day 90 | 15,000 ASCV patients | N/A | [54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Sukhorukov, V.N.; Eremin, I.I.; Nadelyaeva, I.I.; Gutyrchik, N.A.; Orekhov, A.N. Proprotein Convertase Subtilisin/Kexin 9 as a Modifier of Lipid Metabolism in Atherosclerosis. Biomedicines 2023, 11, 503. https://doi.org/10.3390/biomedicines11020503
Poznyak AV, Sukhorukov VN, Eremin II, Nadelyaeva II, Gutyrchik NA, Orekhov AN. Proprotein Convertase Subtilisin/Kexin 9 as a Modifier of Lipid Metabolism in Atherosclerosis. Biomedicines. 2023; 11(2):503. https://doi.org/10.3390/biomedicines11020503
Chicago/Turabian StylePoznyak, Anastasia V., Vasily N. Sukhorukov, Ilya I. Eremin, Irina I. Nadelyaeva, Nikita A. Gutyrchik, and Alexander N. Orekhov. 2023. "Proprotein Convertase Subtilisin/Kexin 9 as a Modifier of Lipid Metabolism in Atherosclerosis" Biomedicines 11, no. 2: 503. https://doi.org/10.3390/biomedicines11020503