
New Monoclonal Antibodies Specific for Different Epitopes of the Spike Protein of SARS-CoV-2 and Its Major Variants: Additional Tools for a More Specific COVID-19 Diagnosis
 , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Expression and Purification of SARS-CoV-2 Proteins in E. coli
2.2. Cloning, Expression and Purification of SARS-CoV-2 Proteins in HEK293T Cells
2.3. N-Deglycosylation of rS1 and rS2 Proteins
2.4. Cells and SARS-CoV-2 Virus Propagation
2.5. SDS-PAGE and WB Analysis
2.6. Mice Immunization
2.7. Isolation and Purification of Specific mAb-Producing Clones
2.8. Nasal Swabs
2.9. ELISA
2.10. Dot Blot Assay
2.11. Lateral Flow Assay (LFA)
2.12. Plaque Reduction Neutralization Test (PRNT)
2.13. Epitope Mapping
2.14. Statistical Analysis
3. Results
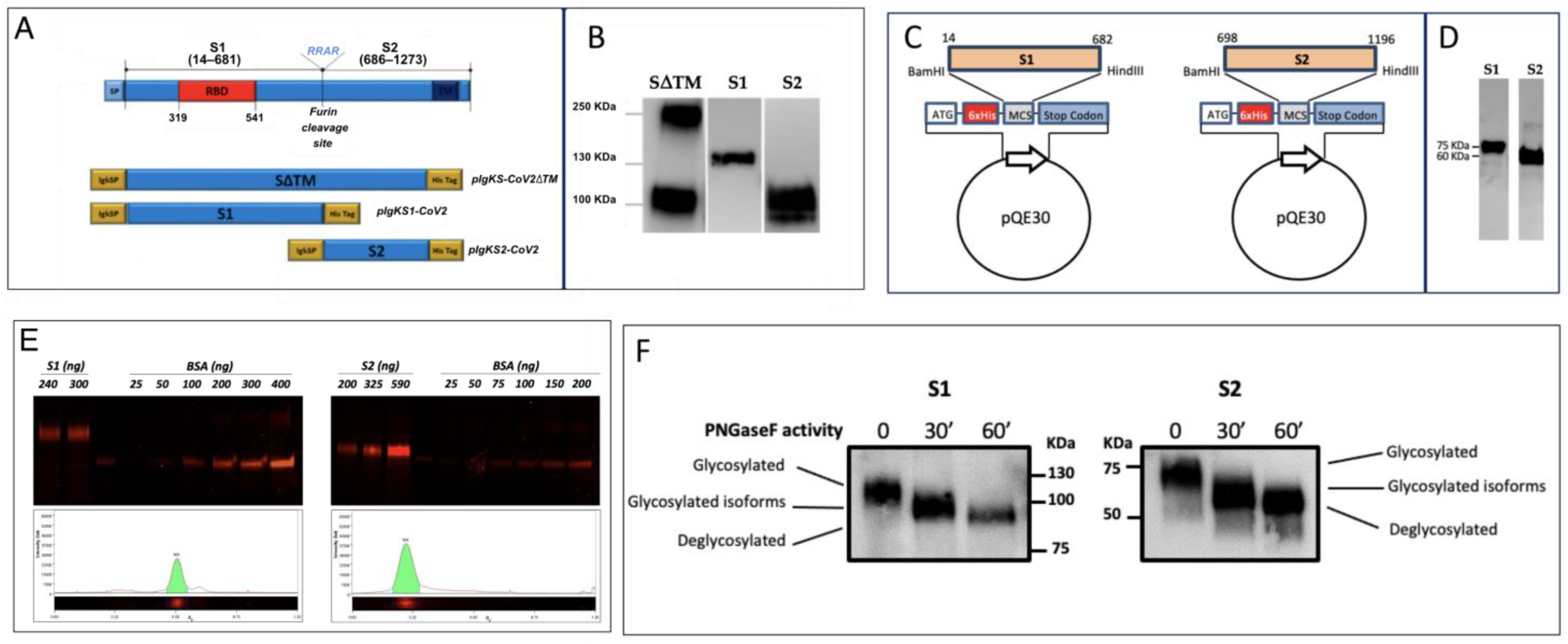
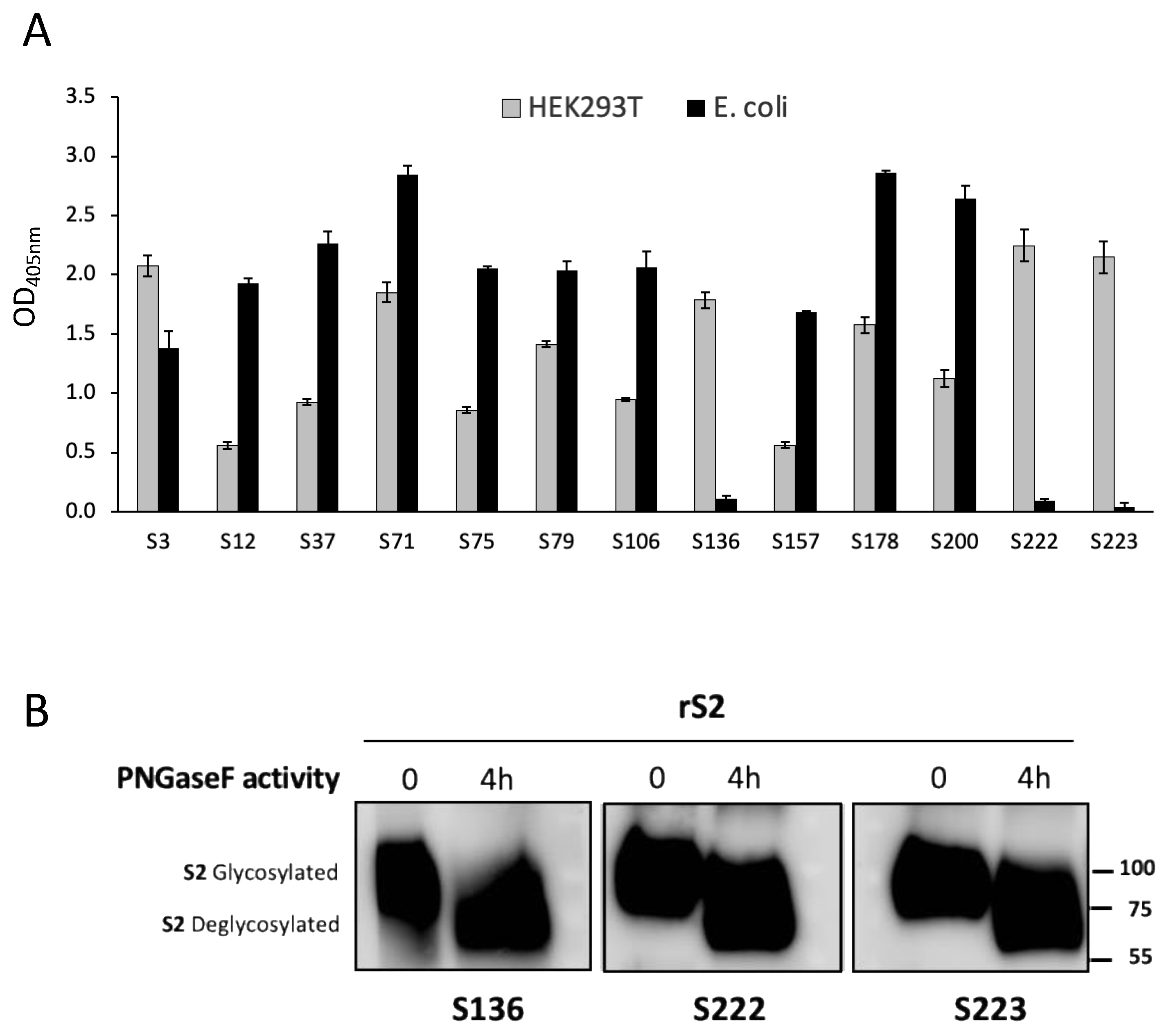
3.1. Production of SARS-CoV-2 Recombinant Proteins
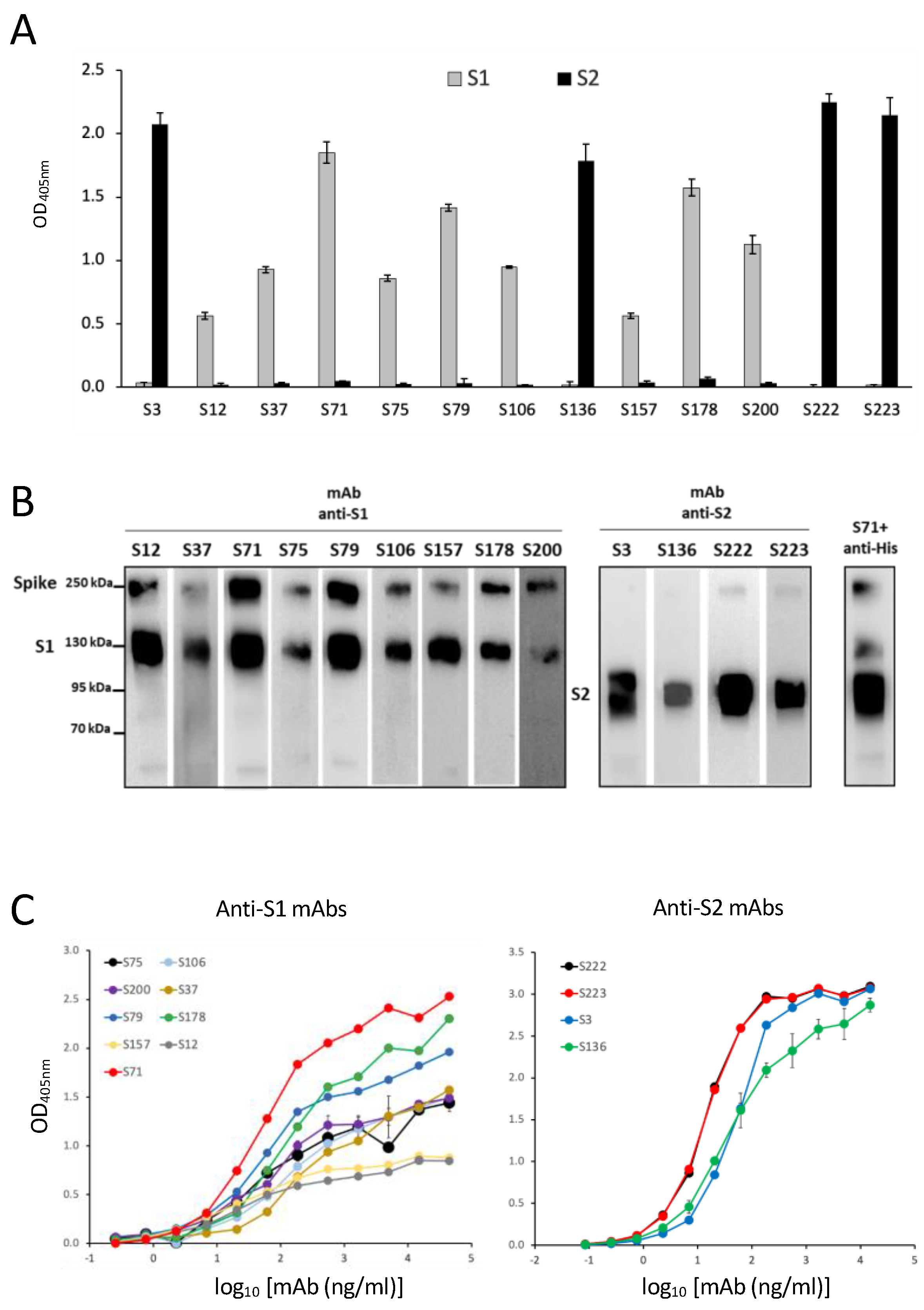
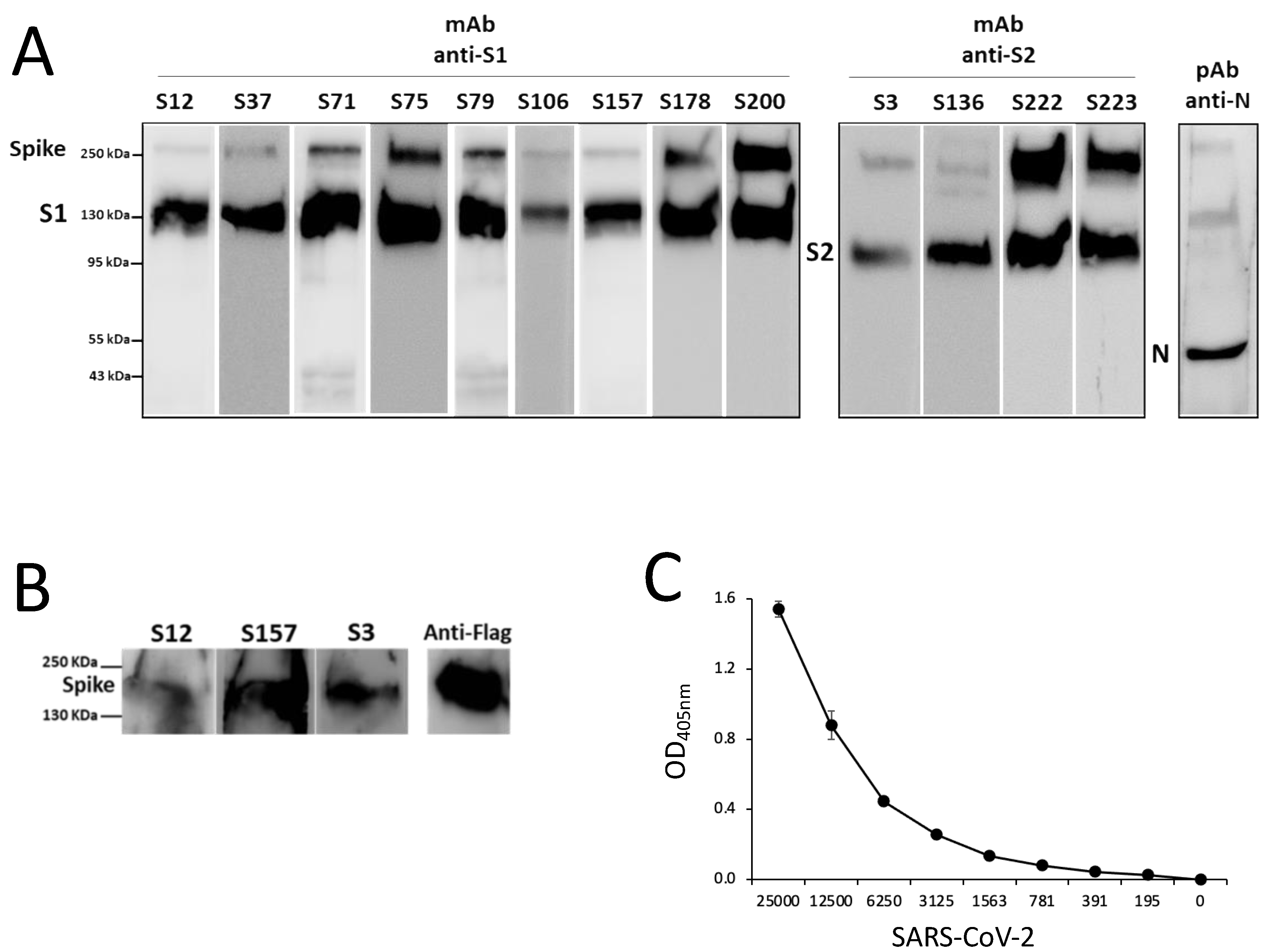
3.2. Production of SARS-CoV-2 Spike-Specific mAbs
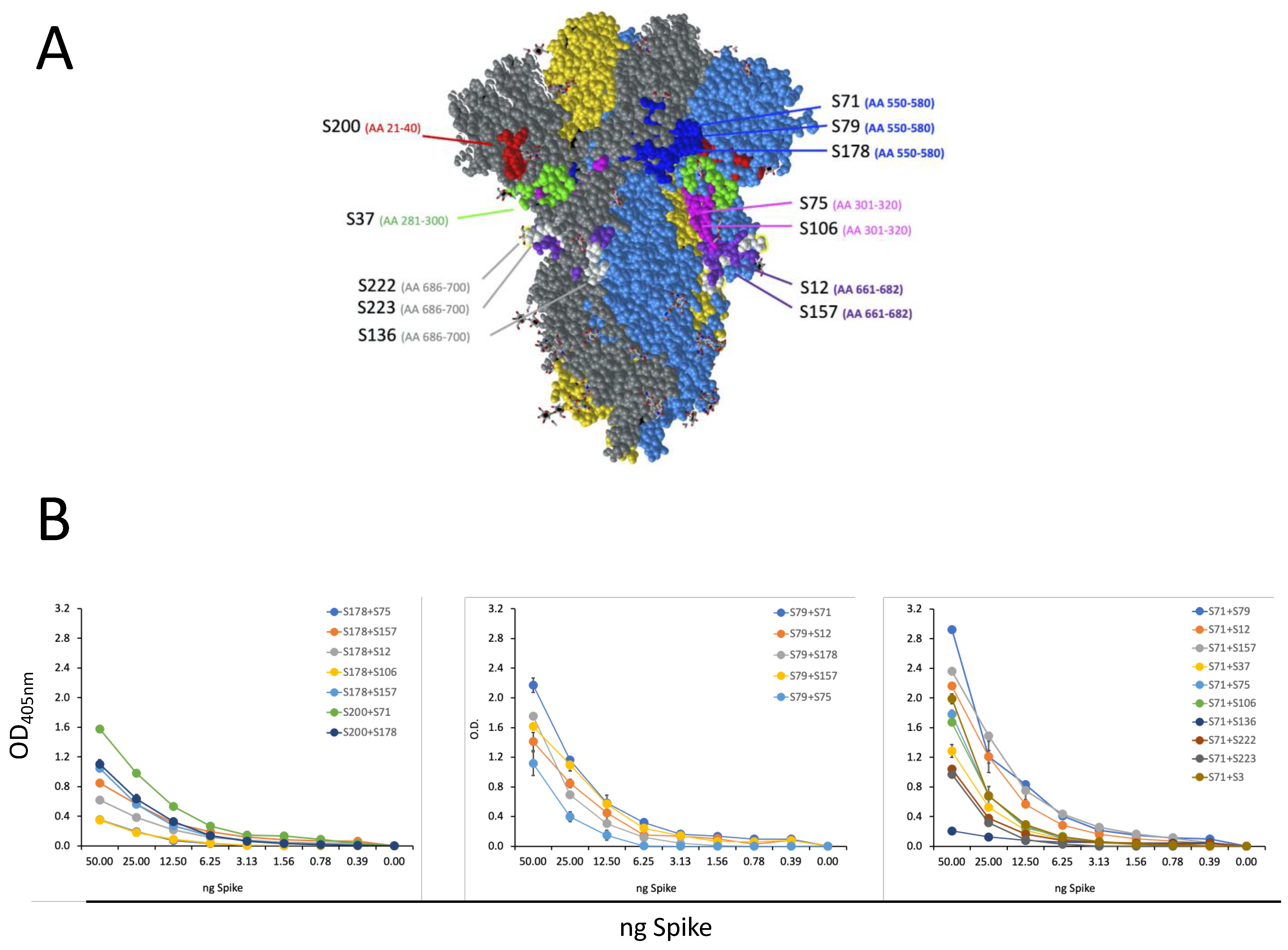
3.3. Characterization of Epitope Recognized by Isolated S-Specific mAbs
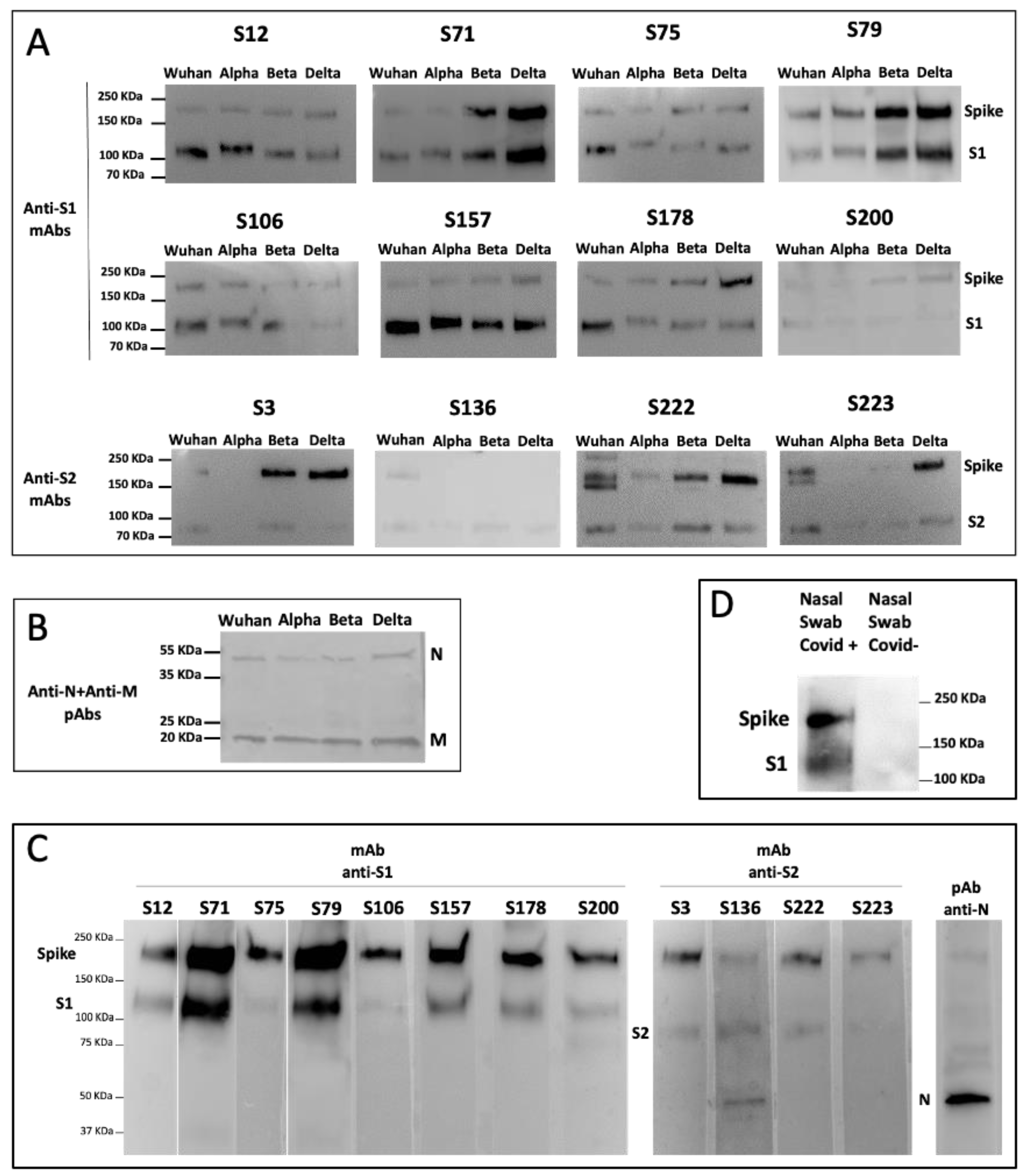
3.4. Isolated S-Specific mAbs Specifically Bind SARS-CoV-2 but no Other Human Coronaviruses
3.5. Isolated S-Specific mAbs Recognize the Major SARS-CoV-2 Variants of Concern
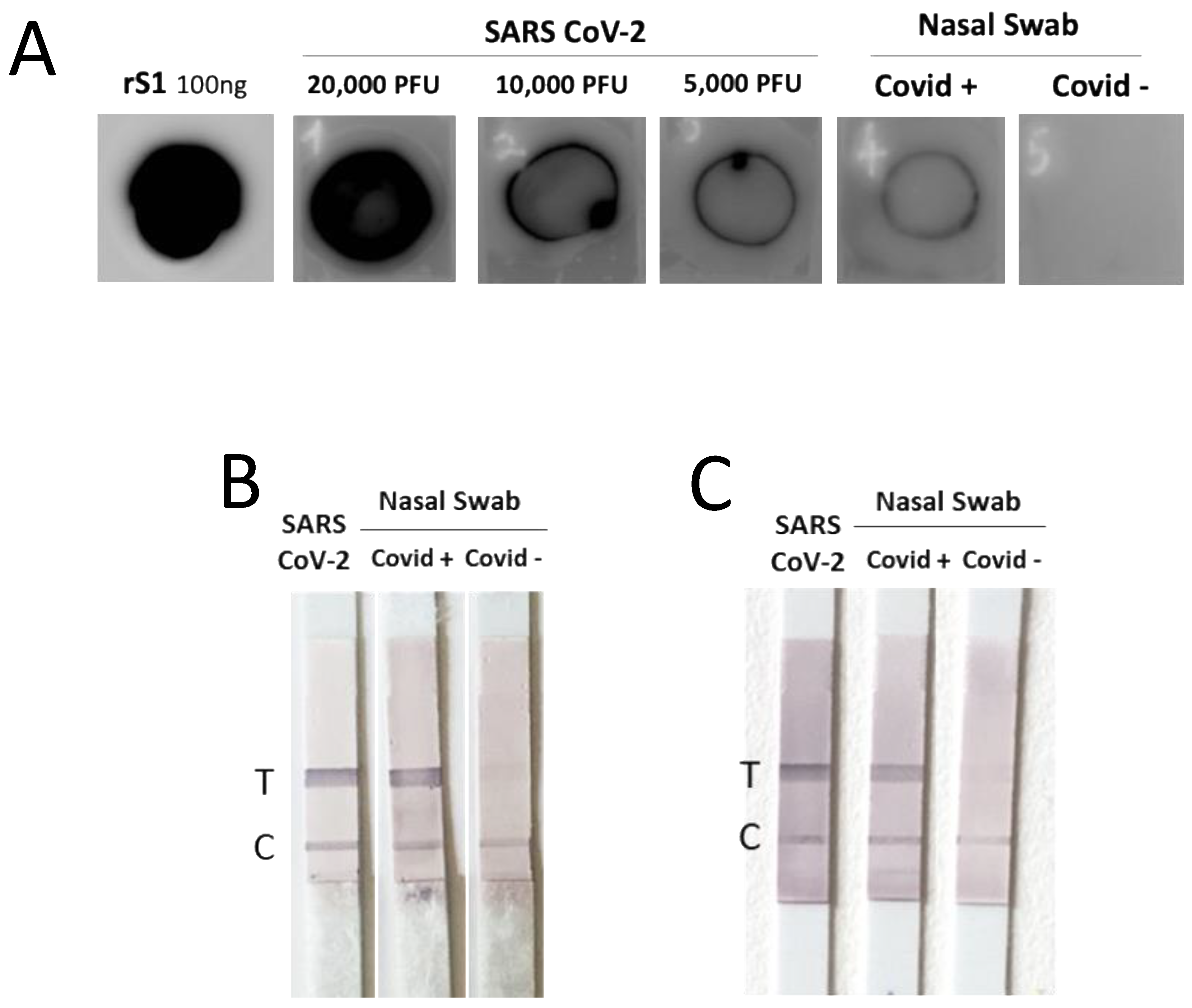
3.6. The Isolated mAbs Are Useful Tools for an Antigenic Diagnostic Assay: A Preliminary Proof-of-Concept
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Abdool Karim, S.S.; de Oliveira, T. New SARS-CoV-2 Variants—Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Limpens, R.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Barcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Zhou, Z.; Huang, C.; Zhou, Z.; Huang, Z.; Su, L.; Kang, S.; Chen, X.; Chen, Q.; He, S.; Rong, X.; et al. Structural insight reveals SARS-CoV-2 ORF7a as an immunomodulating factor for human CD14(+) monocytes. iScience 2021, 24, 102187. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Lieberman, N.A.P.; Phung, Q.; Hsiang, T.Y.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.A.; Huang, M.L.; Gale, M., Jr.; et al. SARS-CoV-2 ORF6 Disrupts Bidirectional Nucleocytoplasmic Transport through Interactions with Rae1 and Nup98. mBio 2021, 12, e00065-21. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.P.; Tang, X.; Lee, J.R.; Chida, A.; Mercer, K.; Wharton, R.E.; Kainulainen, M.; Harcourt, J.L.; Martines, R.B.; Schroeder, M.; et al. Rapid development of neutralizing and diagnostic SARS-COV-2 mouse monoclonal antibodies. Sci. Rep. 2021, 11, 9682. [Google Scholar] [CrossRef]
- Diamond, M.; Chen, R.; Xie, X.; Case, J.; Zhang, X.; VanBlargan, L.; Liu, Y.; Liu, J.; Errico, J.; Winkler, E.; et al. SARS-CoV-2 variants show resistance to neutralization by many monoclonal and serum-derived polyclonal antibodies. Res. Sq. 2021, preprint. [Google Scholar] [CrossRef]
- Chen, R.E.; Winkler, E.S.; Case, J.B.; Aziati, I.D.; Bricker, T.L.; Joshi, A.; Darling, T.L.; Ying, B.; Errico, J.M.; Shrihari, S.; et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature 2021, 596, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Verde, P.; Marcantonio, C.; Costantino, A.; Martina, A.; Simeoni, M.; Taffon, S.; Tritarelli, E.; Campanella, C.; Cresta, R.; Bruni, R.; et al. Diagnostic accuracy of a SARS-CoV-2 rapid antigen test among military and civilian personnel of an Air Force airport in central Italy. PLoS ONE 2022, 17, e0277904. [Google Scholar] [CrossRef] [PubMed]
- Watson, O.J.; Barnsley, G.; Toor, J.; Hogan, A.B.; Winskill, P.; Ghani, A.C. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study. Lancet Infect. Dis. 2022, 22, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, N.; Reddy, A.; Gomez, A.R.; Bosch, I.; Herrera, B.B. Monoclonal antibody pairs against SARS-CoV-2 for rapid antigen test development. PLoS Negl. Trop. Dis. 2022, 16, e0010311. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, N.; Sena, B.F.; Qu, X.; Herrera, B.B. Comparative Evaluation of Rapid Isothermal Amplification and Antigen Assays for Screening Testing of SARS-CoV-2. Viruses 2022, 14, 468. [Google Scholar] [CrossRef] [PubMed]
- Dobano, C.; Santano, R.; Jimenez, A.; Vidal, M.; Chi, J.; Rodrigo Melero, N.; Popovic, M.; Lopez-Aladid, R.; Fernandez-Barat, L.; Tortajada, M.; et al. Immunogenicity and crossreactivity of antibodies to the nucleocapsid protein of SARS-CoV-2: Utility and limitations in seroprevalence and immunity studies. Transl. Res. 2021, 232, 60–74. [Google Scholar] [CrossRef]
- Mariotti, S.; Capocefalo, A.; Chiantore, M.V.; Iacobino, A.; Teloni, R.; De Angelis, M.L.; Gallinaro, A.; Pirillo, M.F.; Borghi, M.; Canitano, A.; et al. Isolation and Characterization of Mouse Monoclonal Antibodies That Neutralize SARS-CoV-2 and Its Variants of Concern Alpha, Beta, Gamma and Delta by Binding Conformational Epitopes of Glycosylated RBD With High Potency. Front. Immunol. 2021, 12, 750386. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 1–16. [Google Scholar] [CrossRef]
- Cox, M.; Peacock, T.P.; Harvey, W.T.; Hughes, J.; Wright, D.W.; Consortium, C.-G.U.; Willett, B.J.; Thomson, E.; Gupta, R.K.; Peacock, S.J.; et al. SARS-CoV-2 variant evasion of monoclonal antibodies based on in vitro studies. Nat. Rev. Microbiol. 2023, 21, 112–124. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Magurano, F.; Baggieri, M.; Marchi, A.; Rezza, G.; Nicoletti, L.; Group, C.-S. SARS-CoV-2 infection: The environmental endurance of the virus can be influenced by the increase of temperature. Clin. Microbiol. Infect. 2021, 27, 289.e5–289.e7. [Google Scholar] [CrossRef] [PubMed]
- Ramirez Hernandez, E.; Hernandez-Zimbron, L.F.; Martinez Zuniga, N.; Leal-Garcia, J.J.; Ignacio Hernandez, V.; Ucharima-Corona, L.E.; Perez Campos, E.; Zenteno, E. The Role of the SARS-CoV-2 S-Protein Glycosylation in the Interaction of SARS-CoV-2/ACE2 and Immunological Responses. Viral Immunol. 2021, 34, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Di Bonito, P.; Grasso, F.; Giorgi, C.; Blasi, F.; Niedrig, M.; Cassone, A. Recombinant protein-based ELISA and immuno-cytochemical assay for the diagnosis of SARS. J. Med. Virol. 2005, 76, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Drain, P.K.; Bemer, M.; Morton, J.F.; Dalmat, R.; Abdille, H.; Thomas, K.K.; Uppal, T.K.; Hau, D.; Green, H.R.; Gates-Hollingsworth, M.A.; et al. Accuracy of 2 Rapid Antigen Tests During 3 Phases of SARS-CoV-2 Variants. JAMA Netw. Open 2022, 5, e2228143. [Google Scholar] [CrossRef] [PubMed]
- Drain, P.K. Rapid Diagnostic Testing for SARS-CoV-2. N. Engl. J. Med. 2022, 386, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Mohit, E.; Rostami, Z.; Vahidi, H. A comparative review of immunoassays for COVID-19 detection. Expert Rev. Clin. Immunol. 2021, 17, 573–599. [Google Scholar] [CrossRef] [PubMed]
- Ng, Q.X.; Lim, Y.L.; Han, M.X.; Teoh, S.E.; Thumboo, J.; Tan, B.H. The Performance of Lateral Flow Tests in the Age of the Omicron: A Rapid Systematic Review. Life 2022, 12, 1941. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, O.; Bergholz, W.; Kisielinski, K.; Giboni, P.; Sonnichsen, A. Methodological problems of SARS-CoV-2 rapid point-of-care tests when used in mass testing. AIMS Public Health 2022, 9, 73–93. [Google Scholar] [CrossRef]
- Kim, T.; Choi, H.; Shin, T.R.; Ko, Y.; Park, Y.B.; Kim, H.I.; Jang, S.H.; Jung, K.S.; Kim, Y.; Lee, M.G.; et al. Epidemiology and clinical features of common community human coronavirus disease. J. Thorac Dis. 2021, 13, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Killerby, M.E.; Biggs, H.M.; Haynes, A.; Dahl, R.M.; Mustaquim, D.; Gerber, S.I.; Watson, J.T. Human coronavirus circulation in the United States 2014-2017. J. Clin. Virol. 2018, 101, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Kozak, R.; Prost, K.; Yip, L.; Williams, V.; Leis, J.A.; Mubareka, S. Severity of coronavirus respiratory tract infections in adults admitted to acute care in Toronto, Ontario. J. Clin. Virol. 2020, 126, 104338. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.Q.; Chen, D.H.; Tan, W.P.; Qiu, S.Y.; Xu, D.; Liang, H.X.; Chen, M.X.; Li, X.; Lin, Z.S.; Liu, W.K.; et al. Epidemiology and clinical characteristics of human coronaviruses OC43, 229E, NL63, and HKU1: A study of hospitalized children with acute respiratory tract infection in Guangzhou, China. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadi, J.; Oueslati, S.; Bellanger, L.; Gallais, F.; Dortet, L.; Roque-Afonso, A.M.; Junot, C.; Naas, T.; Fenaille, F.; Becher, F. Quantitative Assessment of SARS-CoV-2 Virus in Nasopharyngeal Swabs Stored in Transport Medium by a Straightforward LC-MS/MS Assay Targeting Nucleocapsid, Membrane, and Spike Proteins. J. Proteome Res. 2021, 20, 1434–1443. [Google Scholar] [CrossRef]
- Baneyx, F.; Mujacic, M. Recombinant protein folding and misfolding in Escherichia coli. Nat. Biotechnol. 2004, 22, 1399–1408. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, C.; Rao, J.; Chen, L.; Ma, T.; Liu, D.; Ren, L.; Xu, S. The impact of sample processing on the rapid antigen detection test for SARS-CoV-2: Virus inactivation, VTM selection, and sample preservation. Biosaf. Health 2021, 3, 238–243. [Google Scholar] [CrossRef]
- Matricardi, P.M.; Dal Negro, R.W.; Nisini, R. The first, holistic immunological model of COVID-19: Implications for prevention, diagnosis, and public health measures. Pediatr. Allergy Immunol. 2020, 31, 454–470. [Google Scholar] [CrossRef]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct Target Ther. 2021, 6, 396. [Google Scholar] [CrossRef]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers Sequence 5′-3′ | |
|---|---|
| S1 E. coli fw | GCGCGGATCCCAGTGCGTGAACCTGACCACTA |
| S1 E. coli rv | CGCGAAGCTTAGCGAGGGGAGTTAGTCTGGGT |
| S2 E. coli fw | GCGCGGATCCTCACTGGGTGCTGAGAACTCC |
| S2 E. coli rv | GCGCAAGCTTTTAGGACTCGTTCAGGTTCTTGGC |
| S1 HEK fw | GGAAGCTTCGTGAATCTGACAACTCGG |
| S1 HEK rv | GGCTCGAGTCCTTGGAGAGTTTGTCTGGG |
| S2 HEK fw | GGAAGCTTACGGAGCGTGGCATCCCAG |
| S2 HEK rv | GGCTCGAGTCTCCTTCTGGATGTTCACCACGG |
| Peptide # | Amino Acid Position and Sequence |
|---|---|
| 1 | 1 mfvflvllpl vssqcvnltt rtqlppaytn sftrgvyypd 40 |
| 2 | 21 rtqlppaytn sftrgvyypd kvfrssvlhs tqdlflpffs 60 |
| 3 | 41 kvfrssvlhs tqdlflpffs nvtwfhaihv sgtngtkrfd 80 |
| 4 | 61 nvtwfhaihv sgtngtkrfd npvlpfndgv yfasteksni 100 |
| 5 | 81 npvlpfndgv yfasteksni irgwifgttl dsktqslliv 120 |
| 6 | 101 irgwifgttl dsktqslliv nnatnvvikv cefqfcndpf 140 |
| 7 | 121 nnatnvvikv cefqfcndpf lgvyyhknnk swmesefrvy 160 |
| 8 | 141 lgvyyhknnk swmesefrvy ssannctfey vsqpflmdle 180 |
| 9 | 161 ssannctfey vsqpflmdle gkqgnfknlr efvfknidgy 200 |
| 10 | 181 gkqgnfknlr efvfknidgy fkiyskhtpi nlvrdlpqgf 220 |
| 11 | 201 fkiyskhtpi nlvrdlpqgf saleplvdlp iginitrfqt 240 |
| 12 | 221 saleplvdlp iginitrfqt llalhrsylt pgdsssgwta 260 |
| 13 | 241 llalhrsylt pgdsssgwta gaaayyvgyl qprtfllkyn 280 |
| 14 | 261 gaaayyvgyl qprtfllkyn engtitdavd caldplsetk 300 |
| 15 | 281 engtitdavd caldplsetk ctlksftvek giyqtsnfrv 320 |
| 16 | 541 fnfngltgtg vltesnkkfl pfqqfgrdia dttdavrdpq 580 |
| 17 | 561 pfqqfgrdia dttdavrdpq tleilditpc sfggvsvitp 600 |
| 18 | 581 tleilditpc sfggvsvitp gtntsnqvav lyqdvnctev 620 |
| 19 | 601 gtntsnqvav lyqdvnctev pvaihadqlt ptwrvystgs 640 |
| 20 | 621 pvaihadqlt ptwrvystgs nvfqtragcl igaehvnnsy 660 |
| 21 | 641 nvfqtragcl igaehvnnsy ecdipigagi casyqtqtns 680 |
| 22 | 661 ecdipigagi casyqtqtns prrarsvasq siiaytmslg 700 |
| 23 | 681 prrarsvasq siiaytmslg aensvaysnn siaiptnfti 720 |
| 24 | 701 aensvaysnn siaiptnfti svtteilpvs mtktsvdctm 740 |
| mAb | Subclass | Spike Domain | EC50 (ng/mL) | Epitope Position (AA) |
|---|---|---|---|---|
| S200 | IgG1 | S1 | 89.24 | 21–40 |
| S37 | IgG1 | S1 | 301.59 | 281–300 |
| S75 | IgG1 | S1 | 116.71 | 301–320 |
| S106 | IgG1 | S1 | 168.80 | 301–320 |
| S71 | IgG2b | S1 | 86.04 | 550–580 |
| S79 | IgG2a | S1 | 103.81 | 550–580 |
| S178 | IgG2b | S1 | 202.34 | 550–580 |
| S12 | IgG1 | S1 | 66.87 | 661–682 |
| S157 | IgG1 | S1 | 89.32 | 661–682 |
| S222 | IgG1 | S2 | 19.33 | 686–700 |
| S223 | IgG1 | S2 | 18.79 | 686–700 |
| S136 | IgG1 | S2 | 52.32 | 686–700 |
| S3 | IgG1 | S2 | 45.65 | 721–1182 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mariotti, S.; Chiantore, M.V.; Teloni, R.; Iacobino, A.; Capocefalo, A.; Michelini, Z.; Borghi, M.; Baggieri, M.; Marchi, A.; Bucci, P.; et al. New Monoclonal Antibodies Specific for Different Epitopes of the Spike Protein of SARS-CoV-2 and Its Major Variants: Additional Tools for a More Specific COVID-19 Diagnosis. Biomedicines 2023, 11, 610. https://doi.org/10.3390/biomedicines11020610
Mariotti S, Chiantore MV, Teloni R, Iacobino A, Capocefalo A, Michelini Z, Borghi M, Baggieri M, Marchi A, Bucci P, et al. New Monoclonal Antibodies Specific for Different Epitopes of the Spike Protein of SARS-CoV-2 and Its Major Variants: Additional Tools for a More Specific COVID-19 Diagnosis. Biomedicines. 2023; 11(2):610. https://doi.org/10.3390/biomedicines11020610
Chicago/Turabian StyleMariotti, Sabrina, Maria Vincenza Chiantore, Raffaela Teloni, Angelo Iacobino, Antonio Capocefalo, Zuleika Michelini, Martina Borghi, Melissa Baggieri, Antonella Marchi, Paola Bucci, and et al. 2023. "New Monoclonal Antibodies Specific for Different Epitopes of the Spike Protein of SARS-CoV-2 and Its Major Variants: Additional Tools for a More Specific COVID-19 Diagnosis" Biomedicines 11, no. 2: 610. https://doi.org/10.3390/biomedicines11020610
APA StyleMariotti, S., Chiantore, M. V., Teloni, R., Iacobino, A., Capocefalo, A., Michelini, Z., Borghi, M., Baggieri, M., Marchi, A., Bucci, P., Gioacchini, S., D’Amelio, R., Brouwer, P. J. M., Sandini, S., Acchioni, C., Sgarbanti, M., Di Virgilio, A., Grasso, F., Cara, A., ... Nisini, R. (2023). New Monoclonal Antibodies Specific for Different Epitopes of the Spike Protein of SARS-CoV-2 and Its Major Variants: Additional Tools for a More Specific COVID-19 Diagnosis. Biomedicines, 11(2), 610. https://doi.org/10.3390/biomedicines11020610