
Cardioprotective Effects of a Selective c-Jun N-terminal Kinase Inhibitor in a Rat Model of Myocardial Infarction
 , , ,
, , ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Equipment
2.3. Chemicals and Drugs
2.4. Study Compound
2.5. Experimental Protocol
2.6. Model of Myocardium I/R
2.7. Assessment of Cardiac Parameters
2.8. Assessment of Infarct Size
2.9. Morphometric Assessment of Relative Sizes of Scar and LV Cavity and the Entire LV Myocardial Area
2.10. Morphometric Assessment of the Development of Interstitial Fibrosis in the Distant Myocardium (Interventricular Septum)
2.11. Statistical Analysis
3. Results
3.1. Effect of IQ-1 on Survival Rates in Rats 60 Days after MI
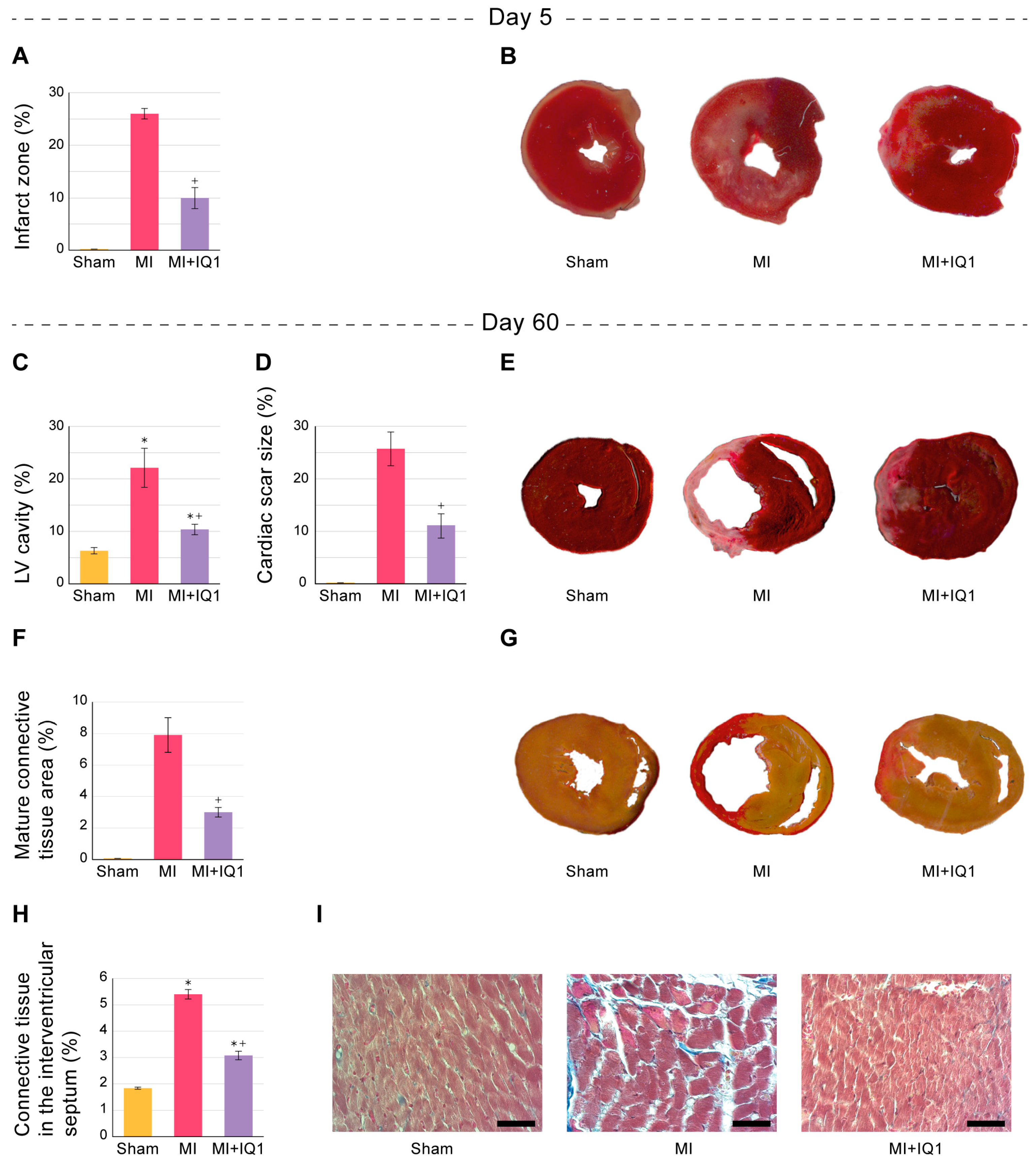
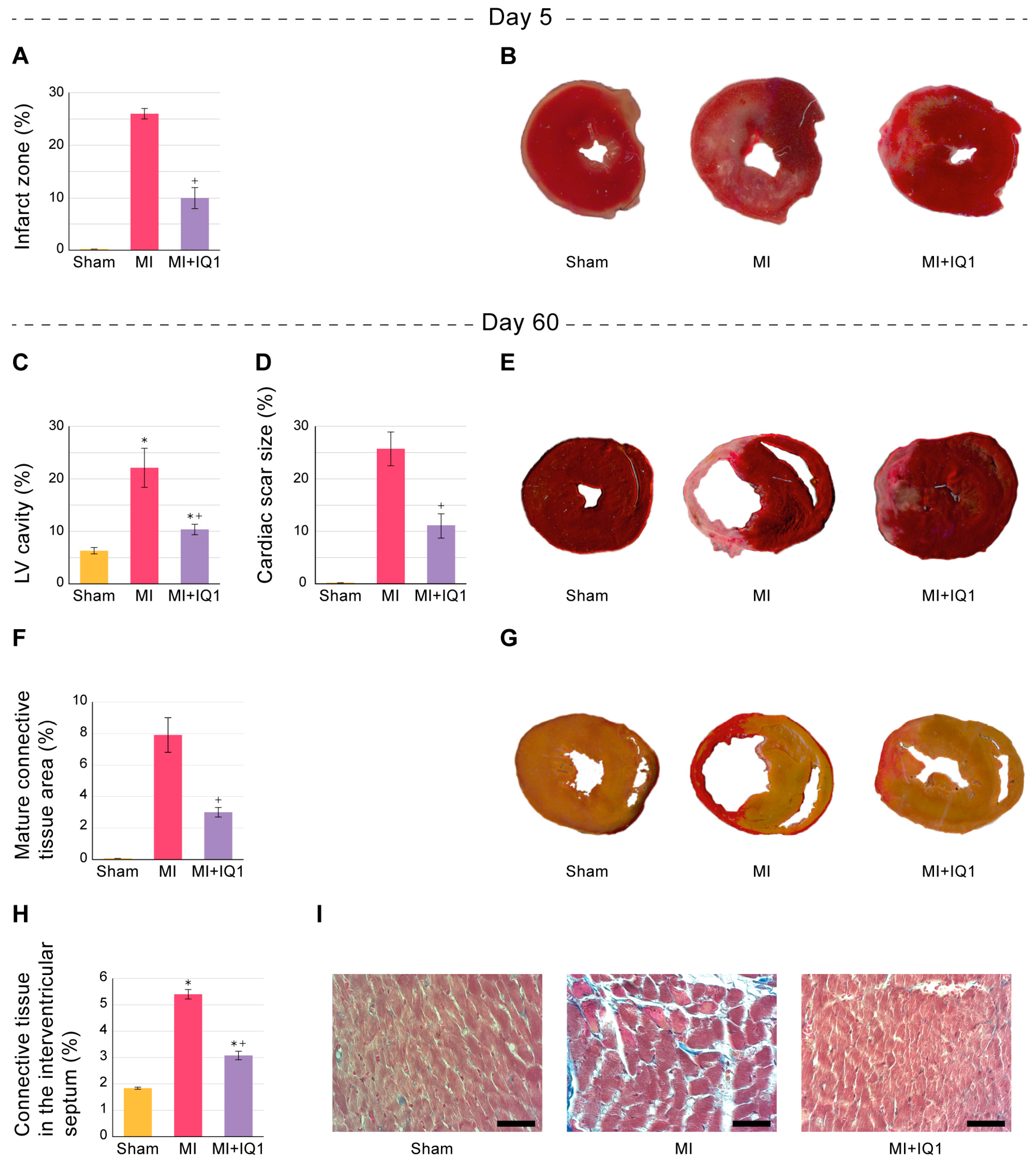
3.2. Effect of IQ-1 on the Size of Infarct Zones in the Myocardium of Rats during the Acute Period after I/R
3.3. Effect of IQ-1 on Systemic Hemodynamic Parameters in Rats 60 Days after MI
3.4. Effect of IQ-1 on Cardiac Contractility Parameters 60 Days after MI
3.5. Effect of IQ-1 on Morphological Changes in Rat Myocardium 60 Days after MI
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reed, G.W.; Rossi, J.E.; Cannon, C.P. Acute myocardial infarction. Lancet 2017, 389, 197–210. [Google Scholar] [CrossRef]
- Lu, Q.; Liu, P.; Huo, J.H.; Wang, Y.N.; Ma, A.Q.; Yuan, Z.Y.; Du, X.J.; Bai, L. Cardiac rupture complicating acute myocardial infarction: The clinical features from an observational study and animal experiment. BMC Cardiovasc. Disord. 2020, 20, 409. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Swaroop, G. Post-myocardial Infarction Heart Failure: A Review on Management of Drug Therapies. Cureus 2022, 14, e25745. [Google Scholar] [CrossRef]
- Nagele, M.P.; Flammer, A.J. Heart Failure After Right Ventricular Myocardial Infarction. Curr. Heart Fail. Rep. 2022, 19, 375–385. [Google Scholar] [CrossRef]
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis Mediates the Decrease in Cellularity during the Transition between Granulation-Tissue and Scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar]
- Sutton, M.G.S.; Sharpe, N. Left ventricular remodeling after myocardial infarction-Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Bolognese, L.; Neskovic, A.N.; Parodi, G.; Cerisano, G.; Buonamici, P.; Santoro, G.M.; Antoniucci, D. Left ventricular remodeling after primary coronary angioplasty-Patterns of left ventricular dilation and long-term prognostic implications. Circulation 2002, 106, 2351–2357. [Google Scholar] [CrossRef] [Green Version]
- Ertl, G.; Frantz, S. Healing after myocardial infarction. Cardiovasc. Res. 2005, 66, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Landmesser, U.; Wollert, K.C.; Drexler, H. Potential novel pharmacological therapies for myocardial remodelling. Cardiovasc. Res. 2009, 81, 519–527. [Google Scholar] [CrossRef]
- Ito, Y.; Ito, K.; Shiroto, T.; Tsuburaya, R.; Yi, G.J.; Takeda, M.; Fukumoto, Y.; Yasuda, S.; Shimokawa, H. Cardiac shock wave therapy ameliorates left ventricular remodeling after myocardial ischemia-reperfusion injury in pigs in vivo. Coron. Artery Dis. 2010, 21, 304–311. [Google Scholar] [CrossRef]
- Murphy, S.P.; Ibrahim, N.E.; Januzzi, J.L., Jr. Heart Failure With Reduced Ejection Fraction: A Review. JAMA 2020, 324, 488–504. [Google Scholar] [CrossRef]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [Green Version]
- Shvedova, M.; Anfinogenova, Y.; Atochina-Vasserman, E.N.; Schepetkin, I.A.; Atochin, D.N. c-Jun N-Terminal Kinases (JNKs) in Myocardial and Cerebral Ischemia/Reperfusion Injury. Front. Pharmacol. 2018, 9, 715. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Li, X.; Zheng, M.Q.; Rozanski, G.J. Role of apoptosis signal-regulating kinase-1-c-Jun NH2-terminal kinase-p38 signaling in voltage-gated K+ channel remodeling of the failing heart: Regulation by thioredoxin. Antioxid. Redox Signal. 2011, 14, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yang, Y.; Xue, C.; Tan, M.; Xu, L.; Gao, J.; Xu, L.; Zong, J.; Qian, W. Zinc Finger Protein ZBTB20 protects against cardiac remodelling post-myocardial infarction via ROS-TNFalpha/ASK1/JNK pathway regulation. J. Cell Mol. Med. 2020, 24, 13383–13396. [Google Scholar] [CrossRef]
- Martinez, P.F.; Bonomo, C.; Guizoni, D.M.; Junior, S.A.; Damatto, R.L.; Cezar, M.D.; Lima, A.R.; Pagan, L.U.; Seiva, F.R.; Bueno, R.T.; et al. Modulation of MAPK and NF-954;B Signaling Pathways by Antioxidant Therapy in Skeletal Muscle of Heart Failure Rats. Cell Physiol. Biochem. 2016, 39, 371–384. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Qi, S.Y.; Ru, L.S.; Ding, C.; Wang, H.J.; Zhao, J.S.; Li, J.J.; Li, A.Y.; Wang, D.M. Reduced Endoplasmic Reticulum Stress Might Alter the Course of Heart Failure Via Caspase-12 and JNK Pathways. Can. J. Cardiol. 2014, 30, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.; Sirker, A.; Shah, A.M. Redox sensitive signaling pathways in cardiac remodeling, hypertrophy and failure. Front. Biosci.-Landmrk 2009, 14, 3168–3187. [Google Scholar] [CrossRef]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009, 81, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, C.J.; Kubasiak, L.A.; Prentice, H.; Andreka, P.; Bishopric, N.H.; Webster, K.A. Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem. J. 2002, 362, 561–571. [Google Scholar] [CrossRef]
- Hreniuk, D.; Garay, M.; Gaarde, W.; Monia, B.P.; McKay, R.A.; Cioffi, C.L. Inhibition of c-Jun N-terminal kinase 1, but not c-Jun N-terminal kinase 2, suppresses apoptosis induced by ischemia/reoxygenation in rat cardiac myocytes. Mol. Pharmacol. 2001, 59, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Pachori, A.; Howard, S.; Iqbal, S.; LoGrasso, P.V. Inhibition of JNK Mitochondrial Localization and Signaling Is Protective against Ischemia/Reperfusion Injury in Rats. J. Biol. Chem. 2013, 288, 4000–4011. [Google Scholar] [CrossRef] [Green Version]
- Duplain, H. Salvage of ischemic myocardium: A focus on JNK. Adv. Exp. Med. Biol. 2006, 588, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, C.; Ballerio, R.; Gaillard, P.; Giachetti, C.; Carboni, S.; Vitte, P.A.; Gotteland, J.P.; Cirillo, R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004, 142, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.H.; Xu, F.F.; Fu, Y.; Liu, F.Y.; Sun, S.; Wu, X.D. Calreticulin induces delayed cardioprotection through mitogen-activated protein kinases. Proteomics 2006, 6, 3792–3800. [Google Scholar] [CrossRef]
- Milano, G.; Morel, S.; Bonny, C.; Samaja, M.; von Segesser, L.K.; Nicod, P.; Vassalli, G. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1828–H1835. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Yao, Y.W.; Su, Z.L.; Yang, Y.B.; Kao, R.; Martin, C.M.; Rui, T. Endogenous HMGB1 contributes to ischemia-reperfusion-induced myocardial apoptosis by potentiating the effect of TNF-alpha/JNK. Am. J. Physiol.-Heart C 2011, 300, H913–H921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, X.X.; Bian, H.J.; Liu, X.B.; Ji, X.P.; Zhang, Y. Inhibition of the activity of Rho-kinase reduces cardiomyocyte apoptosis in heart ischemia/reperfusion via suppressing JNK-mediated AIF translocation. Clin. Chim. Acta 2009, 401, 76–80. [Google Scholar] [CrossRef]
- Schenkel, P.C.; Tavares, A.M.V.; Fernandes, R.O.; Diniz, G.P.; Ludke, A.R.L.; Ribeiro, M.F.M.; Araujo, A.S.D.; Barreto-Chaves, M.L.; Bello-Klein, A. Time course of hydrogen peroxide-thioredoxin balance and its influence on the intracellular signalling in myocardial infarction. Exp. Physiol. 2012, 97, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.C.; Shen, S.T.; Fu, S.Y.; Fang, H.Y.; Wang, X.Z.; Das, S.; Sluijter, J.P.G.; Rosenzweig, A.; Zhou, Y.L.; Kong, X.Q.; et al. Traditional Chinese Medication Qiliqiangxin attenuates cardiac remodeling after acute myocardial infarction in mice. Sci. Rep. 2015, 5, 8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Wei, Z.X.; Zheng, D.D.; Ying, T.; Hong, H.S.; Hu, D.Q.; Lin, Y.L.; Jiang, X.L.; Wu, L.Z.; Lan, T.X.; et al. Recombinant Extracellular Domain (p75ECD) of the Neurotrophin Receptor p75 Attenuates Myocardial Ischemia-Reperfusion Injury by Inhibiting the p-JNK/Caspase-3 Signaling Pathway in Rat Microvascular Pericytes. J. Am. Heart Assoc. 2020, 9, e016047. [Google Scholar] [CrossRef] [PubMed]
- Pando, R.; Cheporko, Y.; Haklai, R.; Maysel-Auslender, S.; Keren, G.; George, J.; Porat, E.; Sagie, A.; Kloog, Y.; Hochhauser, E. Ras inhibition attenuates myocardial ischemia-reperfusion injury. Biochem. Pharmacol. 2009, 77, 1593–1601. [Google Scholar] [CrossRef]
- Gehringer, M.; Muth, F.; Koch, P.; Laufer, S.A. c-Jun N-terminal kinase inhibitors: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 849–872. [Google Scholar] [CrossRef]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of JNKs: Overcoming the dilemma of protection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Hanks, T.S.; Kochetkova, I.; Pascual, D.W.; Jutila, M.A.; Quinn, M.T. Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors. Mol. Pharmacol. 2012, 81, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Pearson, B.D. Indenoquinolines. III. Derivatives of 11H-Indeno-[1,2-b]quinoxaline and related indenoquinolines. J. Org.Chem. 1962, 27, 1674–1678. [Google Scholar] [CrossRef]
- Plotnikova, T.M.; Chernysheva, G.A.; Smol’yakova, V.A.; Shchetinin, P.P.; Kuchin, A.V.; Chukicheva, I.Y.; Plotnikov, M.B. Cardioprotective Activity of 2,6-Diisobornyl-4-Methylphenol in Acute Myocardial Ischemia/Reperfusion in Rats. Bull. Exp. Biol. Med. 2018, 165, 657–659. [Google Scholar] [CrossRef]
- Kogan, A. Surgical method of modeling coronary occlusive infarct and heart aneurysm in rats. Patol. Fiziol. Eksp. Ter. 1979, 3, 79–82. [Google Scholar]
- Cimmino, G.; Ibanez, B.; Giannarelli, C.; Prat-González, S.; Hutter, R.; Garcia, M.; Sanz, J.; Fuster, V.; Badimon, J.J. Carvedilol administration in acute myocardial infarction results in stronger inhibition of early markers of left ventricular remodeling than metoprolol. Int. J. Cardiol. 2011, 153, 256–261. [Google Scholar] [CrossRef]
- Armstrong, S.C. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [Green Version]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, R.J.; Buxton, D.B. Stimulation of c-Jun kinase and mitogen-activated protein kinase by ischemia and reperfusion in the perfused rat heart. Biochem. Biophys. Res. Commun. 1996, 218, 83–88. [Google Scholar] [CrossRef]
- Laderoute, K.R.; Webster, K.A. Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes. Circ. Res. 1997, 80, 336–344. [Google Scholar] [CrossRef]
- He, H.P.; Li, H.L.; Lin, A.N.; Gottlieb, R.A. Activation of the JNK pathway is important for cardiomyocyte death in response to simulated ischemia. Cell Death Differ. 1999, 6, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.M.; Patel, H.H.; Hsu, A.K.; Gross, G.J. Stress-activated protein kinase phosphorylation during cardioprotection in the ischemic myocardium. Am. J. Physiol.-Heart C 2001, 281, H1184–H1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.F.; Ji, X.P.; Li, X.X.; Wang, S.J.; Wang, S.H.; Zhang, Y. Inhibition of the activity of poly (ADP-ribose) polymerase reduces heart ischaemia/reperfusion injury via suppressing JNK-mediated AIF translocation. J. Cell Mol. Med. 2008, 12, 1220–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.D.; Wu, X.; Chen, Q.P.; Zhu, S.S.; Liu, Y.; Pan, D.F.; Chen, X.H.; Li, D.Y. The Anti-Apoptotic and Cardioprotective Effects of Salvianolic Acid A on Rat Cardiomyocytes following Ischemia/Reperfusion by DUSP-Mediated Regulation of the ERK1/2/JNK Pathway. PLoS ONE 2014, 9, e102292. [Google Scholar] [CrossRef]
- Wu, X.; Xu, T.D.; Li, D.Y.; Zhu, S.S.; Chen, Q.P.; Hu, W.J.; Pan, D.F.; Zhu, H.; Sun, H. ERK/PP1a/PLB/SERCA2a and JNK Pathways Are Involved in Luteolin-Mediated Protection of Rat Hearts and Cardiomyocytes following Ischemia/Reperfusion. PLoS ONE 2013, 8, e82957. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; Liu, H.M.; Xie, Z.L.; Yang, S.A.; Xu, W.; Hou, J.B.; Yu, B. Ginsenoside Rb3 Protects Cardiomyocytes against Ischemia-Reperfusion Injury via the Inhibition of JNK-Mediated NF-kappa B Pathway: A Mouse Cardiomyocyte Model. PLoS ONE 2014, 9, e103628. [Google Scholar] [CrossRef]
- Li, C.; Wang, T.; Zhang, C.; Xuan, J.; Su, C.; Wang, Y. Quercetin attenuates cardiomyocyte apoptosis via inhibition of JNK and p38 mitogen-activated protein kinase signaling pathways. Gene 2016, 577, 275–280. [Google Scholar] [CrossRef]
- Shi, S.; Li, Q.S.; Li, H.; Zhang, L.; Xu, M.; Cheng, J.L.; Peng, C.H.; Xu, C.Q.; Tian, Y. Anti-apoptotic action of hydrogen sulfide is associated with early JNK inhibition. Cell Biol. Int. 2009, 33, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Javadov, S. Inhibition of JNK Aggravates the Recovery of Rat Hearts after Global Ischemia: The Role of Mitochondrial JNK. PLoS ONE 2014, 9, e113526. [Google Scholar] [CrossRef] [Green Version]
- Schepetkin, I.A.; Kirpotina, L.N.; Hammaker, D.; Kochetkova, I.; Khlebnikov, A.I.; Lyakhov, S.A.; Firestein, G.S.; Quinn, M.T. Anti-Inflammatory Effects and Joint Protection in Collagen-Induced Arthritis after Treatment with IQ-1S, a Selective c-Jun N-Terminal Kinase Inhibitor. J. Pharmacol. Exp. Ther. 2015, 353, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohit, A.A.; Martin, J.H.; Miller, C.A. p493F12 kinase: A novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 1995, 14, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, M.B.; Chernysheva, G.A.; Smolyakova, V.I.; Aliev, O.I.; Trofimova, E.S.; Sherstoboev, E.Y.; Osipenko, A.N.; Khlebnikov, A.I.; Anfinogenova, Y.J.; Schepetkin, I.A.; et al. Neuroprotective Effects of a Novel Inhibitor of c-Jun N-Terminal Kinase in the Rat Model of Transient Focal Cerebral Ischemia. Cells 2020, 9, 1860. [Google Scholar] [CrossRef] [PubMed]
- Cops, J.; Haesen, S.; De Moor, B.; Mullens, W.; Hansen, D. Current animal models for the study of congestion in heart failure: An overview. Heart Fail. Rev. 2019, 24, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.Y.; Chen, W.W.; Yan, M.J.; Liu, J.S.; Luo, H.L.; Wang, C.; Yang, P. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 2020, 45, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, P.J.F. Pathophysiological Characteristics of the Post-Myocardial Infarction Heart Failure Model in Rats. Arq. Bras. Cardiol. 2011, 96, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Malka, A.; Meerkin, D.; Barac, Y.D.; Malits, E.; Bachner-Hinenzon, N.; Carasso, S.; Ertracht, O.; Angel, I.; Shofti, R.; Youdim, M.; et al. TVP1022: A Novel Cardioprotective Drug Attenuates Left Ventricular Remodeling After Ischemia/Reperfusion in Pigs. J. Cardiovasc. Pharmacol. 2015, 66, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Song, C.L.; Liu, B.; Diao, H.Y.; Shi, Y.F.; Li, Y.X.; Zhang, J.C.; Lu, Y.; Wang, G.; Liu, J.; Yu, Y.P.; et al. The protective effect of microRNA-320 on left ventricular remodeling after myocardial ischemia-reperfusion injury in the rat model. Int. J. Mol. Sci. 2014, 15, 17442–17456. [Google Scholar] [CrossRef]
- Yang, Y.L.; Zhao, L.Y.; Ma, J. Penehyclidine hydrochloride preconditioning provides cardiac protection in a rat model of myocardial ischemia/reperfusion injury via the mechanism of mitochondrial dynamics mechanism. Eur. J. Pharmacol. 2017, 813, 130–139. [Google Scholar] [CrossRef]
- Wu, L.H.; Zhang, Q.; Zhang, S.; Meng, L.Y.; Wang, Y.C.; Sheng, C.J. Effects of gene knockdown of CNP on ventricular remodeling after myocardial ischemia-reperfusion injury through NPRB/Cgmp signaling pathway in rats (Retracted article. See vol. 122, 2021). J. Cell. Biochem. 2018, 119, 1804–1818. [Google Scholar] [CrossRef]
- Dorn, G.W. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc. Res. 2009, 81, 465–473. [Google Scholar] [CrossRef]
- Spinale, F.G.; Mukherjee, R.; Zavadzkas, J.A.; Koval, C.N.; Bouges, S.; Stroud, R.E.; Dobrucki, L.W.; Sinusas, A.J. Cardiac Restricted Overexpression of Membrane Type-1 Matrix Metalloproteinase Causes Adverse Myocardial Remodeling following Myocardial Infarction. J. Biol. Chem. 2010, 285, 30316–30327. [Google Scholar] [CrossRef] [Green Version]
- Aoki, H.; Kang, P.M.; Hampe, J.; Yoshimura, K.; Noma, T.; Matsuzaki, M.; Izumo, S. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J. Biol. Chem. 2002, 277, 10244–10250. [Google Scholar] [CrossRef] [Green Version]
- Engelbrecht, A.-M.; Niesler, C.; Page, C.; Lochner, A. p38 and JNK have distinct regulatory functions on the development of apoptosis during simulated ischaemia and reperfusion in neonatal cardiomyocytes. Basic Res. Cardiol. 2004, 99, 338–350. [Google Scholar] [CrossRef]
- Holmes, J.W.; Borg, T.K.; Covell, J.W. Structure and mechanics of healing myocardial infarcts. Annu. Rev. Biomed. Eng. 2005, 7, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Matsuura, T.R.; Sillje, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ.-Heart Fail. 2021, 14, 112–124. [Google Scholar] [CrossRef]
- Ye, Y.M.; Birnbaum, G.D.; Perez-Polo, J.R.; Nanhwan, M.K.; Nylander, S.; Birnbaum, Y. Ticagrelor Protects the Heart Against Reperfusion Injury and Improves Remodeling After Myocardial Infarction. Arterioscl. Throm. Vas. 2015, 35, 1805–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauss, S.; Schuttler, D.; Bleyer, C.; Vlcek, J.; Shakarami, M.; Tomsits, P.; Schneider, S.; Maderspacher, F.; Chataut, K.; Trebo, A.; et al. Characterization of a porcine model of atrial arrhythmogenicity in the context of ischaemic heart failure. PLoS ONE 2020, 15, e0232374. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.A.; Finato, N.; Rocco, M.; Feruglio, G.A.; Puricelli, C.; Cigola, E.; Quaini, F.; Sonnenblick, E.H.; Olivetti, G.; Anversa, P. Structural Basis of End-Stage Failure in Ischemic Cardiomyopathy in Humans. Circulation 1994, 89, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Volders, P.G.; Willems, I.E.; Cleutjens, J.P.; Arends, J.W.; Havenith, M.G.; Daemen, M.J. Interstitial collagen is increased in the non-infarcted human myocardium after myocardial infarction. J. Mol. Cell. Cardiol. 1993, 25, 1317–1323. [Google Scholar] [CrossRef]
- van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective Effects of a New C-Jun N-terminal Kinase Inhibitor in the Model of Global Cerebral Ischemia in Rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Frazier, D.P.; Wilson, A.; Dougherty, C.J.; Li, H.; Bishopric, N.H.; Webster, K.A. PKC-alpha and TAK-1 are intermediates in the activation of c-Jun NH2-terminal kinase by hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1675–H1684. [Google Scholar] [CrossRef] [PubMed]
- Ventura, J.J.; Cogswell, P.; Flavell, R.A.; Baldwin, A.S., Jr.; Davis, R.J. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 2004, 18, 2905–2915. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal Kinase (JNK) Signaling Initiates Physiological Changes Resulting in Amplification of Reactive Oxygen Species Generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef] [Green Version]
- Khalid, S.; Drasche, A.; Thurner, M.; Hermann, M.; Ashraf, M.I.; Fresser, F.; Baier, G.; Kremser, L.; Lindner, H.; Troppmair, J. cJun N-terminal kinase (JNK) phosphorylation of serine 36 is critical for p66Shc activation. Sci. Rep. 2016, 6, 20930. [Google Scholar] [CrossRef] [Green Version]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, B.; Cimmino, G.; Prat-González, S.; Vilahur, G.; Hutter, R.; García, M.J.; Fuster, V.; Sanz, J.; Badimon, L.; Badimon, J.J. The cardioprotection granted by metoprolol is restricted to its administration prior to coronary reperfusion. Int. J. Cardiol. 2011, 147, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef]
- Seledtsov, V.I.; Malashchenko, V.V.; Meniailo, M.E.; Atochin, D.N.; Seledtsova, G.V.; Schepetkin, I.A. Inhibitory effect of IQ-1S, a selective c-Jun N-terminal kinase (JNK) inhibitor, on phenotypical and cytokine-producing characteristics in human macrophages and T-cells. Eur. J. Pharmacol. 2020, 878, 173116. [Google Scholar] [CrossRef]
- Lin, X.; Jo, H.; Ishii, T.M.; Fujita, M.; Fu, M.; Tambara, K.; Yamamoto, M.; Tabata, Y.; Komeda, M.; Matsuoka, S. Controlled release of matrix metalloproteinase-1 plasmid DNA prevents left ventricular remodeling in chronic myocardial infarction of rats. Circ. J. 2009, 73, 2315–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Zhu, W.; Wan, D. Downregulation of microRNA-21-5p from macrophages-derived exosomes represses ventricular remodeling after myocardial infarction via inhibiting tissue inhibitors of metalloproteinase 3. Int. Immunopharmacol. 2021, 96, 107611. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Parameter | Group | ||
|---|---|---|---|
| Sham-Operated (n = 7) | Control (n = 11) | IQ-1 (n = 11) | |
| Stroke volume (mL) | 0.22 ± 0.02 | 0.15 ± 0.01 * | 0.20 ± 0.01 # |
| Heart rate (min–1) | 381 ± 19 | 383 ± 10 | 393 ± 14 |
| Cardiac output (mL × min–1) | 84.8 ± 6.1 | 56.4 ± 3.0 * | 78.7 ± 3.2 # |
| Contractility index (s–1) | 119 ± 1 | 79 ± 3 * | 106 ± 2 # |
| Aortal pressure (mm Hg) | 119 ± 3 | 101 ± 3 * | 113 ± 3 # |
| Left ventricular systolic pressure (mm Hg) | 129 ± 4 | 108 ± 7 * | 122 ± 3 # |
| +dP/dtmax (mm Hg/s) | 7566 ± 124 | 4870 ± 363 * | 6585 ± 172 # |
| –dP/dtmax (mm Hg/s) | –6369 ± 152 | –3295 ± 263 * | –4875 ± 160 # |
| Left ventricular end-diastolic pressure (mm Hg) | 1.4 ± 1.1 | 13.2 ± 1.8 * | 3.9 ± 0.9 # |
| Minimum pressure in the left ventricle (mm Hg) | –1.8 ± 1.1 | 10.0 ± 1.7 * | 0.8 ± 1.0 # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plotnikov, M.B.; Chernysheva, G.A.; Smol’yakova, V.I.; Aliev, O.I.; Fomina, T.I.; Sandrikina, L.A.; Sukhodolo, I.V.; Ivanova, V.V.; Osipenko, A.N.; Anfinogenova, N.D.; et al. Cardioprotective Effects of a Selective c-Jun N-terminal Kinase Inhibitor in a Rat Model of Myocardial Infarction. Biomedicines 2023, 11, 714. https://doi.org/10.3390/biomedicines11030714
Plotnikov MB, Chernysheva GA, Smol’yakova VI, Aliev OI, Fomina TI, Sandrikina LA, Sukhodolo IV, Ivanova VV, Osipenko AN, Anfinogenova ND, et al. Cardioprotective Effects of a Selective c-Jun N-terminal Kinase Inhibitor in a Rat Model of Myocardial Infarction. Biomedicines. 2023; 11(3):714. https://doi.org/10.3390/biomedicines11030714
Chicago/Turabian StylePlotnikov, Mark B., Galina A. Chernysheva, Vera I. Smol’yakova, Oleg I. Aliev, Tatyana I. Fomina, Lyubov A. Sandrikina, Irina V. Sukhodolo, Vera V. Ivanova, Anton N. Osipenko, Nina D. Anfinogenova, and et al. 2023. "Cardioprotective Effects of a Selective c-Jun N-terminal Kinase Inhibitor in a Rat Model of Myocardial Infarction" Biomedicines 11, no. 3: 714. https://doi.org/10.3390/biomedicines11030714