
Michael Adduct of Sulfonamide Chalcone Targets Folate Metabolism in Brugia Malayi Parasite

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
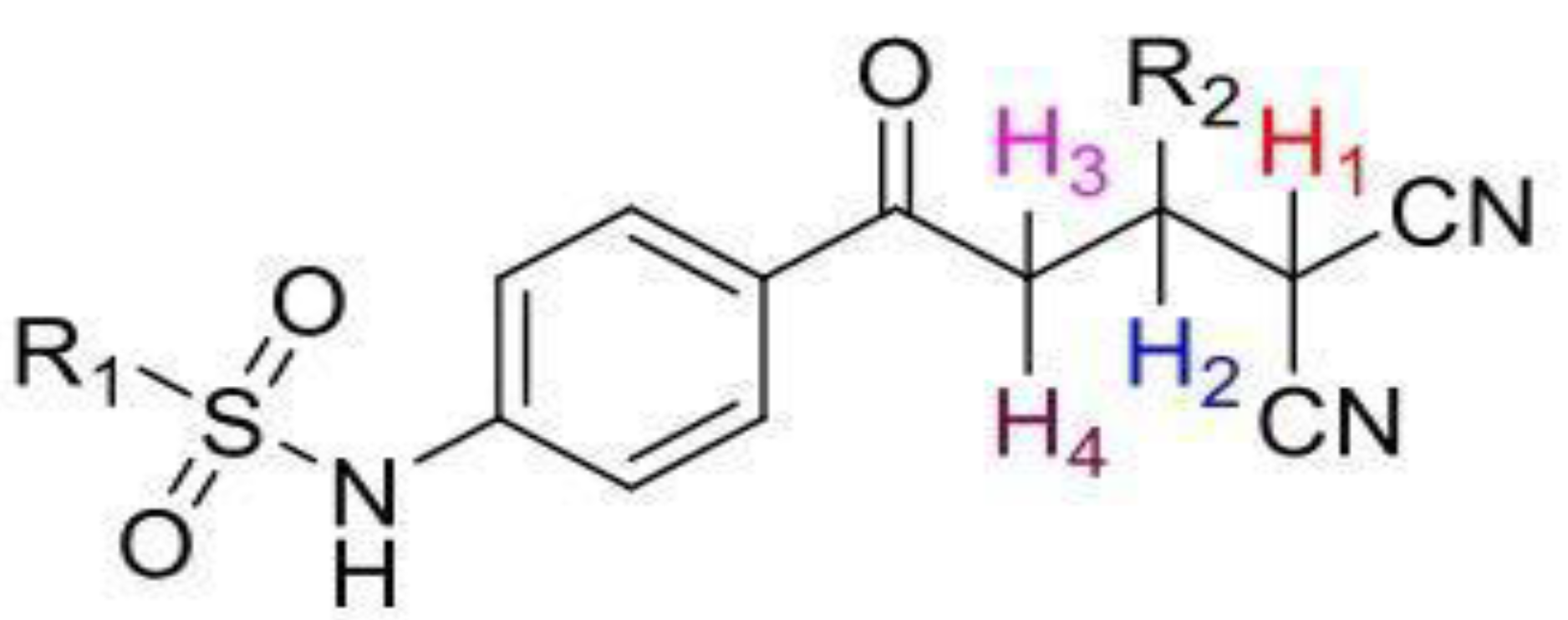
2.1. Chemistry
2.1.1. General Method of Synthesis
(4-(4,4-dicyano-3-phenylbutanoyl)phenyl)benzenesulfonamide (3a)
N-(4-(4,4-dicyano-3-(4-methoxyphenyl)butanoyl)phenyl)benzenesulfonamide (3b)
N-(4-(3-(4-chlorophenyl)-4,4-dicyanobutanoyl)phenyl)benzenesulfonamide (3c)
N-(4-(3-(4-bromophenyl)-4,4-dicyanobutanoyl)phenyl)benzenesulfonamide (3d)
N-(4-(4,4-dicyano-3-(3,4,5-trimethoxyphenyl)butanoyl)phenyl)benzene-sulfonamide (3e)
N-(4-(3-(2-chlorophenyl)-4,4-dicyanobutanoyl)phenyl)benzenesulfonamide (3f)
N-(4-(4,4-dicyano-3-p-tolylbutanoyl)phenyl)benzenesulfonamide (3g)
N-(4-(4,4-dicyano-3-(4-methoxyphenyl)butanoyl)phenyl)4-methylbenzene-sulfonamide (3h)
N-(4-(3-(4-chlorophenyl)-4,4-dicyanobutanoyl)phenyl)-4-methylbenzene-sulfonamide (3i)
N-(4-(3-(4-bromophenyl)-4,4-dicyanobutanoyl)phenyl)4-methylbenzene-sulfonamide (3j)
N-(4-(4,4-dicyano-3-(4-isopropylphenyl)butanoyl)phenyl)-4-methylbenzene-sulfonamide (3k)
N-(4-(3-(2-chlorophenyl)-4,4-dicyanobutanoyl)phenyl)-4-methylbenzene-sulfonamide (3l)
N-(4-(4,4-dicyano-3-p-tolylbutanoyl)phenyl)-4-methylbenzenesulfonamide (3m)
N-(4-(3-(3-chlorophenyl)-4,4-dicyanobutanoyl)phenyl)-4-methylbenzene-sulfonamide (3n)
2.1.2. Biological Activity
Microfilariae Collection
In Vitro Screening of Compounds for Antifilarial Activity
Determination of Lethal Dose of Chalcone Derivatives
Molecular Docking Studies
DHFR Enzyme Assay
Folate Reversal Studies
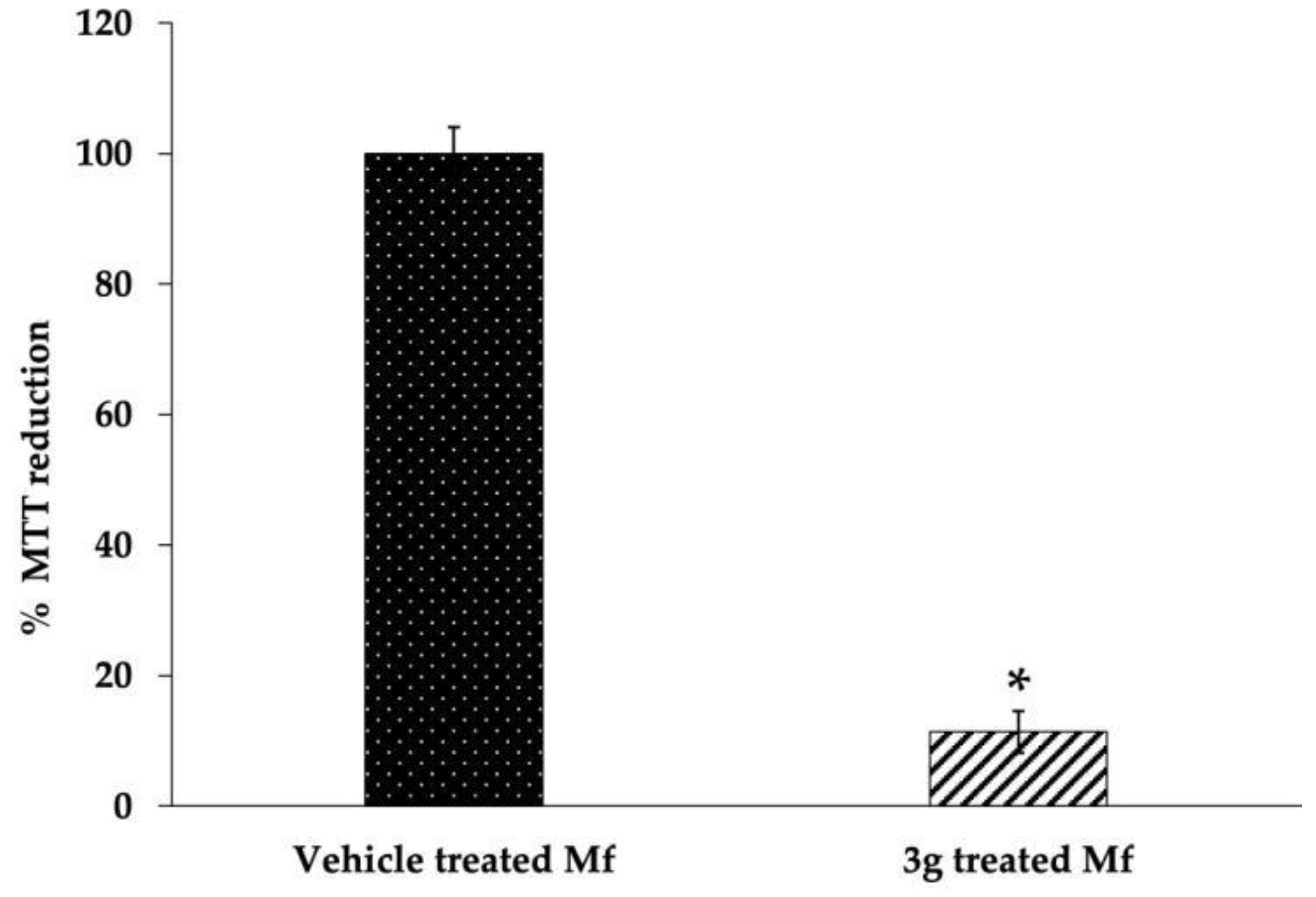
MTT Assay
Acridine Orange–Ethidium Bromide (AO/EB) Staining for Determination of Apoptosis
Cytochrome c ELISA
2.1.3. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
3.2. Biology
3.3. Antifilarial Activity and Cytotoxicity
3.4. DHFR as a Target
3.5. Induction of Apoptosis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Lymphatic Filariasis. 2022. Available online: https://www.who.int/health-topics/lymphatic-filariasis#tab=tab_1 (accessed on 1 December 2022).
- Kulkarni, P.; Thomas, J.J.; Dowerah, J.; Narayana Murthy, M.R.; Ravikumar, K. Mass Drug Administration Programme against Lymphatic Filariasis-an Evaluation of Coverage and Compliance in a Northern Karnataka District, India. Clin. Epidemiol. Glob. Health 2020, 8, 87–90. [Google Scholar] [CrossRef] [Green Version]
- Yajima, A.; Ichimori, K. Progress in the Elimination of Lymphatic Filariasis in the Western Pacific Region: Successes and Challenges. Int. Health 2020, 13, S10–S16. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.; Roy, N.; Dhingra, N. Introduction of Triple-Drug Therapy for Accelerating Lymphatic Filariasis Elimination in India: Lessons Learned. Am. J. Trop. Med. Hyg. 2022, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Behm, C.A.; Bendig, M.M.; McCarter, J.P.; Sluder, A.E. WHO/TDR Scientific Working Group on “RNA Interference as a Means of Identifying Drug Targets for Filariasis Report. 2004. Available online: https://www.semanticscholar.org/paper/For-the-Web-Only-Who%2Ftdr-Scientific-Working-Group-a-Behm-Bendig/e3cd5111be68b7591b9e12a21d75405aa68cccd3 (accessed on 1 December 2022).
- Gupta, S.; Srivastava, A.K. Biochemical Targets in Filarial Worms for Selective Antifilarial Drug Design. Acta Parasitol. 2005, 1, 1–8. [Google Scholar]
- Peixoto, C.A.; Santos, A.C.O.; Ayres, C.F.J. Molecular Evidence for Apoptosis in Microfilariae of Wuchereria bancrofti Induced by Diethylcarbamazine. Parasitol. Res. 2008, 103, 717–721. [Google Scholar] [CrossRef]
- Sharma, R.; Petare, S.; Shinde, G.; Kalyan, G.; Reddy, M. Novel Drug Designing Rationale against Brugia malayi Microfilariae Using Herbal Extracts. Asian Pac. J. Trop. Med. 2010, 3, 846–850. [Google Scholar] [CrossRef] [Green Version]
- Tzin, V.; Galili, G. The Biosynthetic Pathways for Shikimate and Aromatic Amino Acids in Arabidopsis thaliana. Arab. Book 2010, 8, e0132. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Perán, E.; Cabezas-Herrera, J.; García-Cánovas, F.; Durrant, M.C.; Thorneley, R.N.F.; Rodríguez-López, J.N. The Antifolate Activity of Tea Catechins. Cancer Res. 2005, 65, 2059–2064. [Google Scholar] [CrossRef] [Green Version]
- K Sahu, N.; S Balbhadra, S.; Choudhary, J.; V Kohli, D. Exploring Pharmacological Significance of Chalcone Scaffold: A Review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef]
- Singh, A.; Dixit, S.K.; Yadav, M.; Yadav, S.S.; Rathaur, S.; Awasthi, S.K.; Mishra, N. Antifilarial Activity of 1,3-Diarylpropen-1-One: Effect on Glutathione-S-Transferase, a Phase II Detoxification Enzyme. Am. J. Trop. Med. Hyg. 2009, 80, 764–768. [Google Scholar] [CrossRef] [Green Version]
- Bahekar, S.P.; Hande, S.V.; Agrawal, N.R.; Chandak, H.S.; Bhoj, P.S.; Goswami, K.; Reddy, M.V.R. Sulfonamide Chalcones: Synthesis and in Vitro Exploration for Therapeutic Potential against Brugia malayi. Eur. J. Med. Chem. 2016, 124, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Sänger, I.; Lämmler, G.; Kimmig, P. Filarial Infections of Mastomys Natalensis and Their Relevance for Experimental Chemotherapy. Acta Trop. 1981, 38, 277–288. [Google Scholar]
- Bhoj, P.S.; Rao, S.; Bahekar, S.P.; Agrawal, N.R.; Togre, N.S.; Sharma, R.; Goswami, K.; Chandak, H.S.; Patil, M.B. In Vitro Apoptotic Effect on Human Lymphatic Filarial Parasite by Piperidine Derivatives and Thymidine Reversal Study. Parasitol. Res. 2020, 119, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Hande, S.; Goswami, K.; Sharma, R.; Bhoj, P.; Jena, L.; Reddy, M.V.R. Targeting Folate Metabolism for Therapeutic Option: A Bioinformatics Approach. Indian J. Exp. Biol. 2015, 53, 762–766. [Google Scholar] [PubMed]
- Widemann, B.C.; Balis, F.M.; Adamson, P.C. Dihydrofolate Reductase Enzyme Inhibition Assay for Plasma Methotrexate Determination Using a 96-Well Microplate Reader. Clin. Chem. 1999, 45, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, U.R.; Mehta, K.; Subrahmanyam, D.; Vickery, A.C. Brugia malayi and Acanthocheilonema viteae: Antifilarial Activity of Transglutaminase Inhibitors in Vitro. Antimicrob. Agents Chemother. 1991, 35, 2219–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sashidhara, K.V.; Rao, K.B.; Kushwaha, V.; Modukuri, R.K.; Verma, R.; Murthy, P.K. Synthesis and Antifilarial Activity of Chalcone–Thiazole Derivatives against a Human Lymphatic Filarial Parasite, Brugia malayi. Eur. J. Med. Chem. 2014, 81, 473–480. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Khazir, J.; Riley, D.L.; Chashoo, G.; Mir, B.A.; Liles, D.; Islam, M.A.; Singh, S.K.; Vishwakarma, R.A.; Pilcher, L.A. Design, Synthesis and Anticancer Activity of Michael-Type Thiol Adducts of α-Santonin Analogue with Exocyclic Methylene. Eur. J. Med. Chem. 2015, 101, 769–779. [Google Scholar] [CrossRef]
- Brogden, R.N.; Carmine, A.A.; Heel, R.C.; Speight, T.M.; Avery, G.S. Domperidone. Drugs 1982, 24, 360–400. [Google Scholar] [CrossRef]
- Hagner, N.; Joerger, M. Cancer Chemotherapy: Targeting Folic Acid Synthesis. Cancer Manag. Res. 2010, 2, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Bennuru, S.; Meng, Z.; Ribeiro, J.M.C.; Semnani, R.T.; Ghedin, E.; Chan, K.; Lucas, D.A.; Veenstra, T.D.; Nutman, T.B. Stage-Specific Proteomic Expression Patterns of the Human Filarial Parasite Brugia malayi and Its Endosymbiont Wolbachia. Proc. Natl. Acad. Sci. USA 2011, 108, 9649–9654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Abraham, R.; Sanchez, K.G.; Alfonso, M.; Gubler, U.; Siekierka, J.J.; Goodey, N.M. Expression, Purification and Enzymatic Characterization of Brugia malayi Dihydrofolate Reductase. Protein Expr. Purif. 2016, 128, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Tobias, A.M.; Toska, D.; Lange, K.; Eck, T.; Bhat, R.; Janson, C.A.; Rotella, D.P.; Gubler, U.; Goodey, N.M. Expression, Purification, and Inhibition Profile of Dihydrofolate Reductase from the Filarial Nematode Wuchereria bancrofti. PLoS ONE 2018, 13, e0197173. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
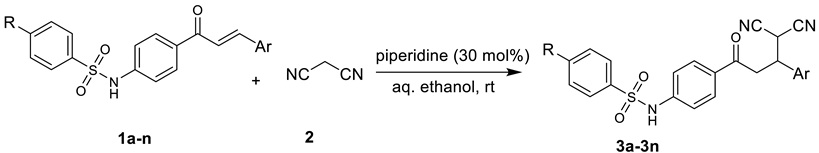 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Ar | Product | Time (min) | Yield (%) | Mp (°C) |
| 3a | H | -Ph |  | 15 | 94 | 179–180 |
| 3b | H | -4-MeO-C6H4 |  | 20 | 81 | 191–193 |
| 3c | H | -4-Cl-C6H4 |  | 5 | 87 | 182–183 |
| 3d | H | -4-Br-C6H4 |  | 5 | 82 | 210–212 |
| 3e | H | -4-(MeO)3-C6H2 |  | 30 | 96 | 187–188 |
| 3f | H | -2-Cl-C6H4 | 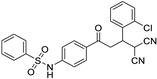 | 5 | 75 | 181–182 |
| 3g | H | -4-CH3-C6H4 | 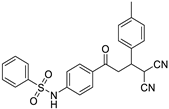 | 20 | 70 | 191–192 |
| 3h | -CH3 | -4-MeO-C6H4 | 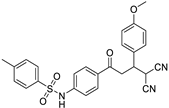 | 15 | 84 | 162–164 |
| 3i | -CH3 | -4-Cl-C6H4 |  | 5 | 86 | 178–179 |
| 3j | -CH3 | -4-Br-C6H4 | 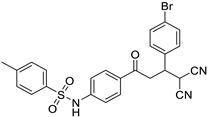 | 5 | 88 | 192–194 |
| 3k | -CH3 | -3-iPr-C6H4 |  | 25 | 80 | 175–176 |
| 3l | -CH3 | -2-Cl-C6H4 |  | 5 | 85 | 182–183 |
| 3m | -CH3 | -4-CH3-C6H4 | 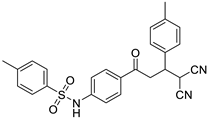 | 20 | 77 | 167–168 |
| 3n | -CH3 | -3-Cl-C6H4 |  | 5 | 90 | 183–184 |
| Compound | Chalcone | Michael Adduct (MA) |
|---|---|---|
| Structure |  | 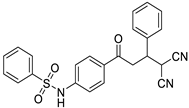 |
| Free energy of binding (∆Gb) | −6.93 kcal/Mol | −9.46 kcal/Mol |
| Inhibition constant (Ki) | 8.31 µM | 115.69 nM |
| Hydrogen bonding | Gly116:HN::O:Chalcone | Gly116:HN::OS:MA |
| Compound | IC100 |
|---|---|
| 3c | 114 ± 9 μM |
| 3g | 38 ± 1 μM |
| 3i | 132 ± 4 μM |
| 3l | 210 ± 0 μM |
| Compound | ∆Gb | Ki | Hydrogen Bonds |
|---|---|---|---|
| 3c | −9.58 kcal/Mol | 94.52 nM | No H-bond formed |
| 3g | −9.54 kcal/Mol | 101.83 nM | NH of Leu29 of BmDHFR: sulfonyl group (O=S=O) of 3g |
| 3i | −9.87 kcal/Mol | 58.61 nM | No H-bond formed |
| 3l | −9.41 kcal/Mol | 125.65 nM | No H-bond formed |
| ECG | −7.82 kcal/Mol | 1.84 µM | NH of Leu29 of BmDHFR: H of ECG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhoj, P.S.; Bahekar, S.P.; Chowdhary, S.; Togre, N.S.; Amdare, N.P.; Jena, L.; Goswami, K.; Chandak, H. Michael Adduct of Sulfonamide Chalcone Targets Folate Metabolism in Brugia Malayi Parasite. Biomedicines 2023, 11, 723. https://doi.org/10.3390/biomedicines11030723
Bhoj PS, Bahekar SP, Chowdhary S, Togre NS, Amdare NP, Jena L, Goswami K, Chandak H. Michael Adduct of Sulfonamide Chalcone Targets Folate Metabolism in Brugia Malayi Parasite. Biomedicines. 2023; 11(3):723. https://doi.org/10.3390/biomedicines11030723
Chicago/Turabian StyleBhoj, Priyanka S., Sandeep P. Bahekar, Shambhavi Chowdhary, Namdev S. Togre, Nitin P. Amdare, Lingaraj Jena, Kalyan Goswami, and Hemant Chandak. 2023. "Michael Adduct of Sulfonamide Chalcone Targets Folate Metabolism in Brugia Malayi Parasite" Biomedicines 11, no. 3: 723. https://doi.org/10.3390/biomedicines11030723